Hartmann Stefanie, Vision Todd J
Department of Biology, University of North Carolina, Chapel Hill, NC 27599, USA.
BMC Evol Biol. 2008 Mar 26;8:95. doi: 10.1186/1471-2148-8-95.
While full genome sequences are still only available for a handful of taxa, large collections of partial gene sequences are available for many more. The alignment of partial gene sequences results in a multiple sequence alignment containing large gaps that are arranged in a staggered pattern. The consequences of this pattern of missing data on the accuracy of phylogenetic analysis are not well understood. We conducted a simulation study to determine the accuracy of phylogenetic trees obtained from gappy alignments using three commonly used phylogenetic reconstruction methods (Neighbor Joining, Maximum Parsimony, and Maximum Likelihood) and studied ways to improve the accuracy of trees obtained from such datasets.
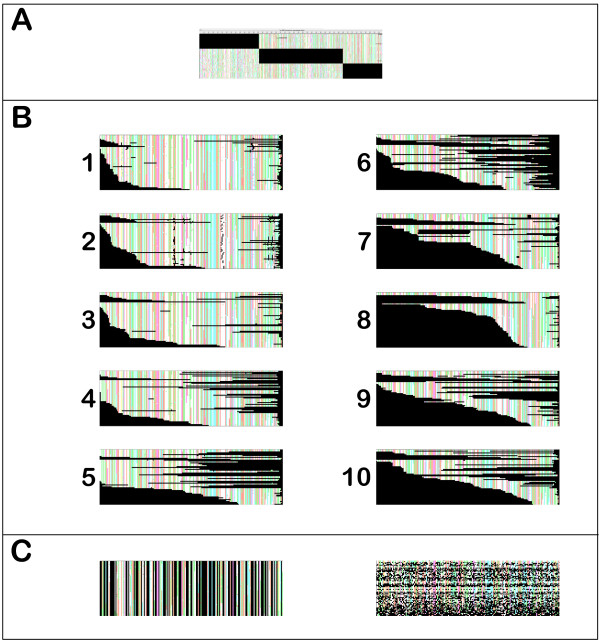
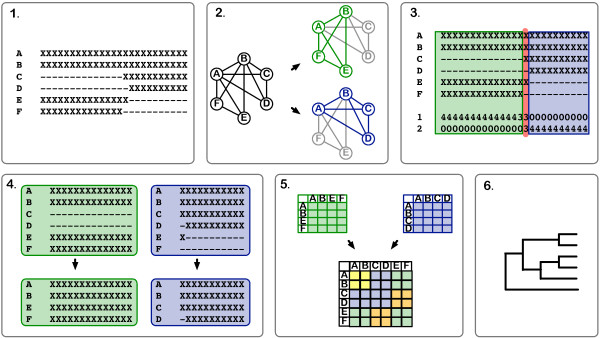
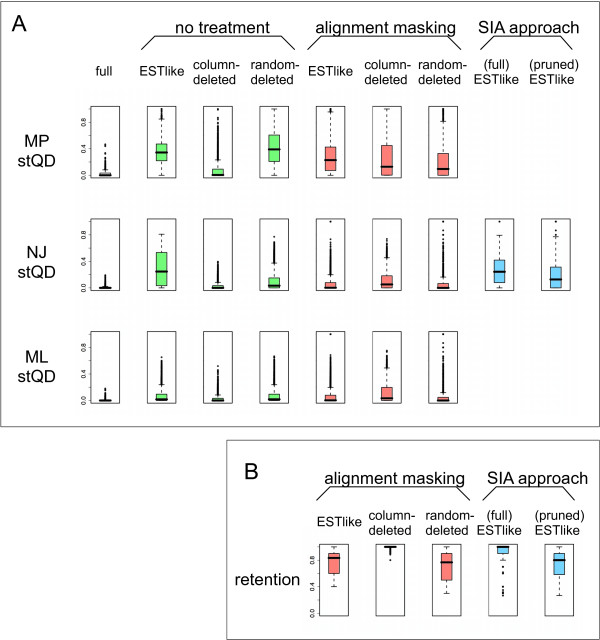
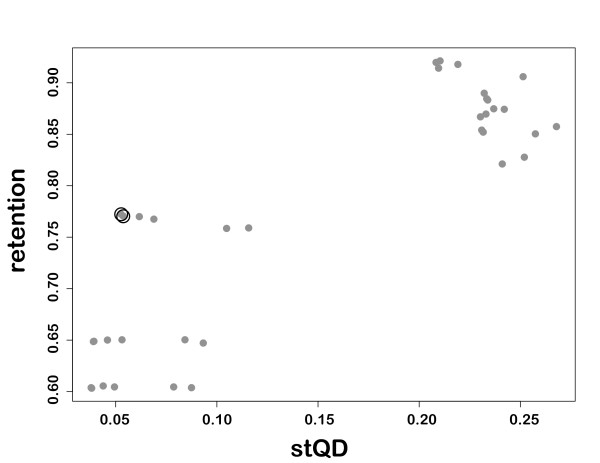
We found that the pattern of gappiness in multiple sequence alignments derived from partial gene sequences substantially compromised phylogenetic accuracy even in the absence of alignment error. The decline in accuracy was beyond what would be expected based on the amount of missing data. The decline was particularly dramatic for Neighbor Joining and Maximum Parsimony, where the majority of gappy alignments contained 25% to 40% incorrect quartets. To improve the accuracy of the trees obtained from a gappy multiple sequence alignment, we examined two approaches. In the first approach, alignment masking, potentially problematic columns and input sequences are excluded from from the dataset. Even in the absence of alignment error, masking improved phylogenetic accuracy up to 100-fold. However, masking retained, on average, only 83% of the input sequences. In the second approach, alignment subdivision, the missing data is statistically modelled in order to retain as many sequences as possible in the phylogenetic analysis. Subdivision resulted in more modest improvements to alignment accuracy, but succeeded in including almost all of the input sequences.
These results demonstrate that partial gene sequences and gappy multiple sequence alignments can pose a major problem for phylogenetic analysis. The concern will be greatest for high-throughput phylogenomic analyses, in which Neighbor Joining is often the preferred method due to its computational efficiency. Both approaches can be used to increase the accuracy of phylogenetic inference from a gappy alignment. The choice between the two approaches will depend upon how robust the application is to the loss of sequences from the input set, with alignment masking generally giving a much greater improvement in accuracy but at the cost of discarding a larger number of the input sequences.
虽然目前仅有少数分类单元拥有完整的基因组序列,但更多分类单元拥有大量的部分基因序列集合。部分基因序列的比对会产生一个包含大量以交错模式排列的缺口的多序列比对。这种缺失数据模式对系统发育分析准确性的影响尚未得到充分理解。我们进行了一项模拟研究,以确定使用三种常用的系统发育重建方法(邻接法、最大简约法和最大似然法)从有缺口的比对中获得的系统发育树的准确性,并研究提高从此类数据集中获得的树的准确性的方法。
我们发现,即使在没有比对错误的情况下,由部分基因序列产生的多序列比对中的缺口模式也会严重损害系统发育准确性。准确性的下降超出了基于缺失数据量所预期的范围。对于邻接法和最大简约法,准确性下降尤为显著,其中大多数有缺口的比对包含25%至40%的错误四重奏。为了提高从有缺口的多序列比对中获得的树的准确性,我们研究了两种方法。在第一种方法,即比对屏蔽中,将潜在有问题的列和输入序列从数据集中排除。即使在没有比对错误的情况下,屏蔽也能将系统发育准确性提高多达100倍。然而,屏蔽平均仅保留83%的输入序列。在第二种方法,即比对细分中,对缺失数据进行统计建模,以便在系统发育分析中保留尽可能多的序列。细分对比对准确性的提高较为有限,但成功地纳入了几乎所有的输入序列。
这些结果表明,部分基因序列和有缺口的多序列比对可能给系统发育分析带来重大问题。对于高通量系统发育基因组学分析,这种担忧最为突出,因为邻接法因其计算效率常常是首选方法。两种方法都可用于提高从有缺口的比对中进行系统发育推断的准确性。两种方法的选择将取决于应用对输入集中序列丢失的稳健程度,比对屏蔽通常能在准确性上有更大提升,但代价是丢弃更多的输入序列。