Wong Thomas K F, Kalyaanamoorthy Subha, Meusemann Karen, Yeates David K, Misof Bernhard, Jermiin Lars S
Land & Water, CSIRO, Canberra, ACT 2601, Australia.
Research School of Biology, Australian National University, Canberra, ACT 2600, Australia.
NAR Genom Bioinform. 2020 Apr 14;2(2):lqaa024. doi: 10.1093/nargab/lqaa024. eCollection 2020 Jun.
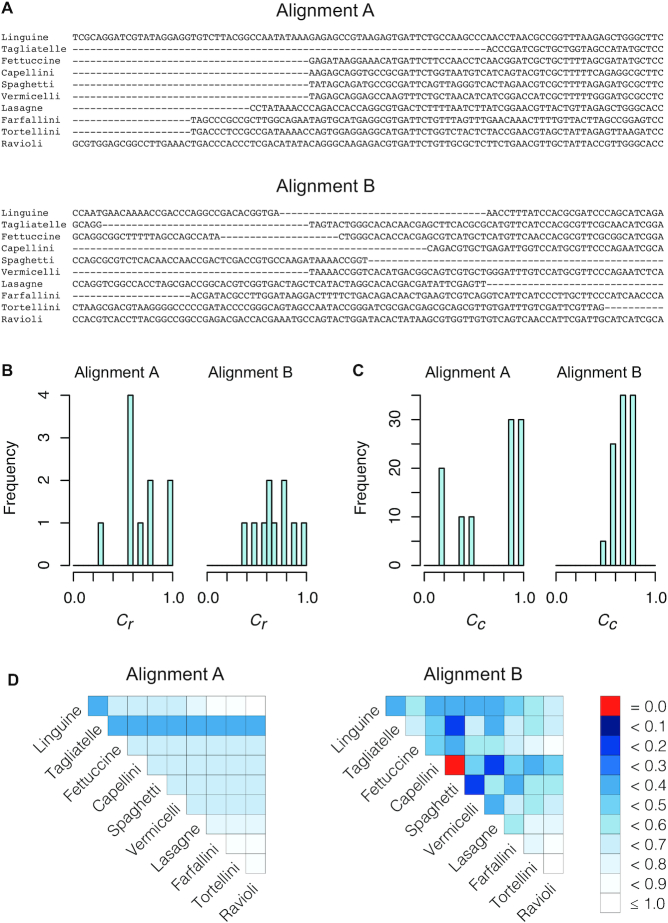
Multiple sequence alignments (MSAs) play a pivotal role in studies of molecular sequence data, but nobody has developed a minimum reporting standard (MRS) to quantify the completeness of MSAs in terms of completely specified nucleotides or amino acids. We present an MRS that relies on four simple completeness metrics. The metrics are implemented in AliStat, a program developed to support the MRS. A survey of published MSAs illustrates the benefits and unprecedented transparency offered by the MRS.
多序列比对(MSA)在分子序列数据研究中起着关键作用,但尚未有人制定最低报告标准(MRS),以便根据完全指定的核苷酸或氨基酸来量化MSA的完整性。我们提出了一种基于四个简单完整性指标的MRS。这些指标在AliStat中得以实现,AliStat是为支持该MRS而开发的程序。对已发表的MSA进行的一项调查说明了该MRS所带来的益处和前所未有的透明度。