Zhang Xu, Shiu Shin-Han, Cal Andrew, Borevitz Justin O
Department of Ecology and Evolution, University of Chicago, Chicago, Illinois, United States of America.
PLoS Genet. 2008 Mar 21;4(3):e1000032. doi: 10.1371/journal.pgen.1000032.
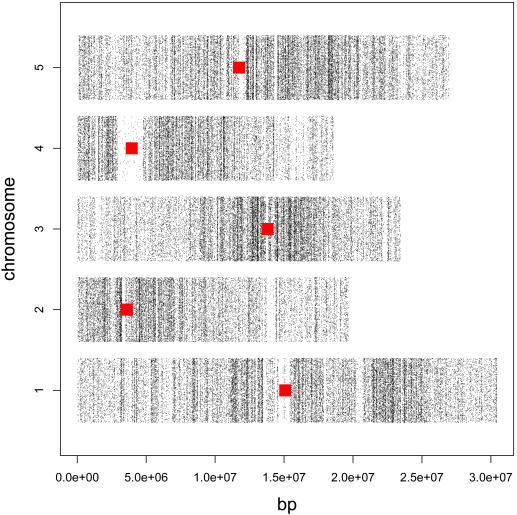
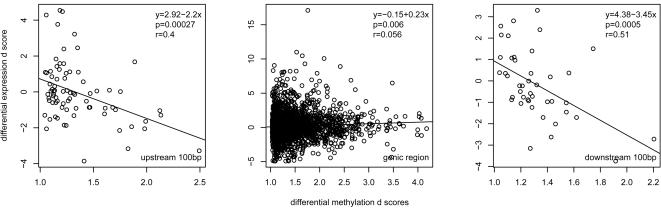
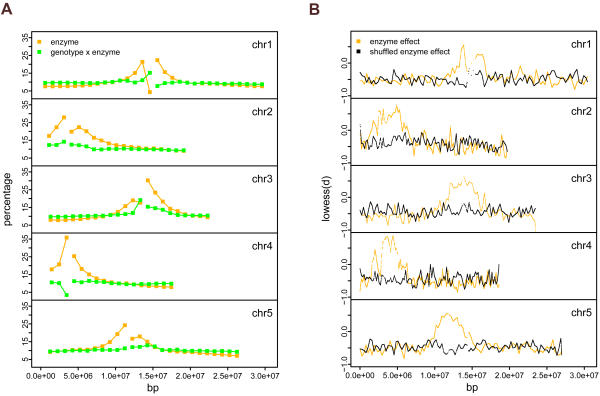
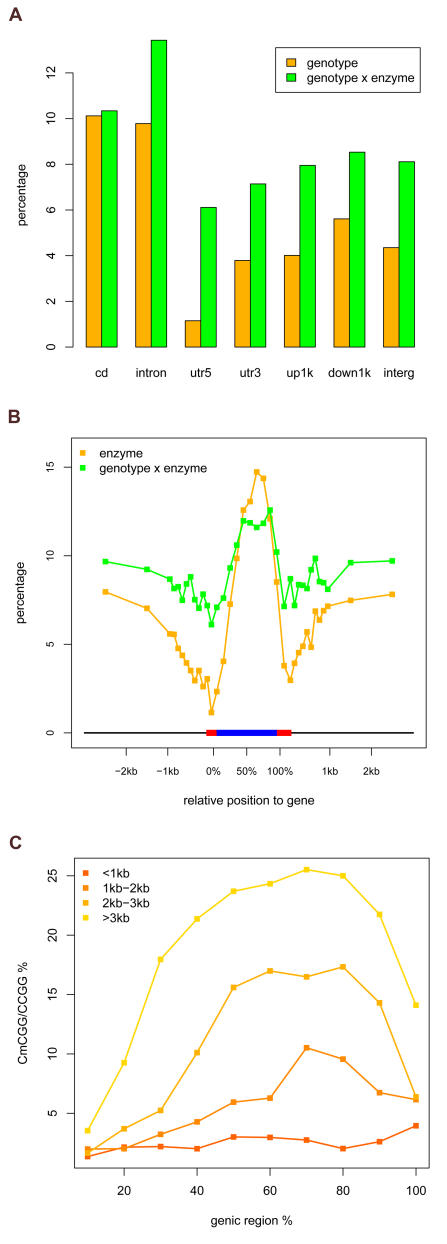
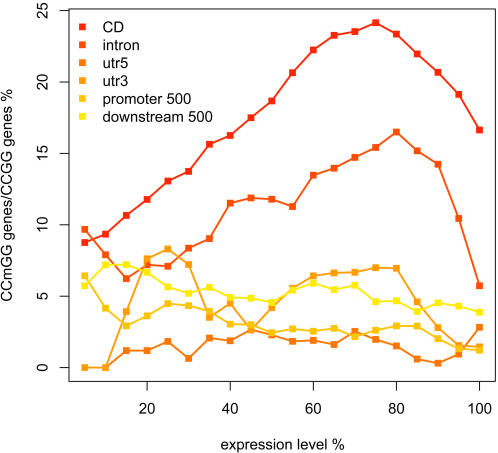
Whole genome tiling arrays provide a high resolution platform for profiling of genetic, epigenetic, and gene expression polymorphisms. In this study we surveyed natural genomic variation in cytosine methylation among Arabidopsis thaliana wild accessions Columbia (Col) and Vancouver (Van) by comparing hybridization intensity difference between genomic DNA digested with either methylation-sensitive (HpaII) or -insensitive (MspI) restriction enzyme. Single Feature Polymorphisms (SFPs) were assayed on a full set of 1,683,620 unique features of Arabidopsis Tiling Array 1.0F (Affymetrix), while constitutive and polymorphic CG methylation were assayed on a subset of 54,519 features, which contain a 5'CCGG3' restriction site. 138,552 SFPs (1% FDR) were identified across enzyme treatments, which preferentially accumulated in pericentromeric regions. Our study also demonstrates that at least 8% of all analyzed CCGG sites were constitutively methylated across the two strains, while about 10% of all analyzed CCGG sites were differentially methylated between the two strains. Within euchromatin arms, both constitutive and polymorphic CG methylation accumulated in central regions of genes but under-represented toward the 5' and 3' ends of the coding sequences. Nevertheless, polymorphic methylation occurred much more frequently in gene ends than constitutive methylation. Inheritance of methylation polymorphisms in reciprocal F1 hybrids was predominantly additive, with F1 plants generally showing levels of methylation intermediate between the parents. By comparing gene expression profiles, using matched tissue samples, we found that magnitude of methylation polymorphism immediately upstream or downstream of the gene was inversely correlated with the degree of expression variation for that gene. In contrast, methylation polymorphism within genic region showed weak positive correlation with expression variation. Our results demonstrated extensive genetic and epigenetic polymorphisms between Arabidopsis accessions and suggested a possible relationship between natural CG methylation variation and gene expression variation.
全基因组平铺阵列提供了一个高分辨率平台,用于分析遗传、表观遗传和基因表达多态性。在本研究中,我们通过比较用甲基化敏感(HpaII)或甲基化不敏感(MspI)限制性内切酶消化的基因组DNA之间的杂交强度差异,调查了拟南芥野生生态型哥伦比亚(Col)和温哥华(Van)之间胞嘧啶甲基化的自然基因组变异。在拟南芥平铺阵列1.0F(Affymetrix)的全套1,683,620个独特特征上检测单特征多态性(SFP),而在包含5'CCGG3'限制性位点的54,519个特征的子集上检测组成型和多态性CG甲基化。在酶处理之间鉴定出138,552个SFP(FDR为1%),这些SFP优先聚集在着丝粒周围区域。我们的研究还表明,在两个菌株中,至少8%的所有分析的CCGG位点是组成型甲基化的,而在两个菌株之间,约10%的所有分析的CCGG位点是差异甲基化的。在常染色质臂内,组成型和多态性CG甲基化都聚集在基因的中心区域,但在编码序列的5'和3'末端代表性不足。然而,多态性甲基化在基因末端比组成型甲基化更频繁地发生。在正反交F1杂种中,甲基化多态性的遗传主要是加性的,F1植株通常表现出介于亲本之间的甲基化水平。通过使用匹配的组织样本比较基因表达谱,我们发现基因上游或下游紧邻区域的甲基化多态性程度与该基因的表达变异程度呈负相关。相比之下,基因区域内的甲基化多态性与表达变异呈弱正相关。我们的结果证明了拟南芥不同生态型之间存在广泛的遗传和表观遗传多态性,并表明自然CG甲基化变异与基因表达变异之间可能存在关系。