Watson Melanie R, Lagow Robert D, Xu Kexiang, Zhang Bing, Bonini Nancy M
Department of Biology, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA.
J Biol Chem. 2008 Sep 5;283(36):24972-81. doi: 10.1074/jbc.M804817200. Epub 2008 Jul 2.
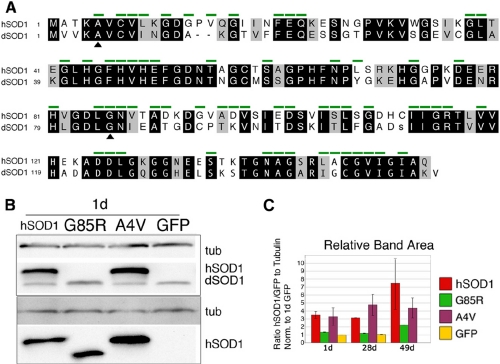
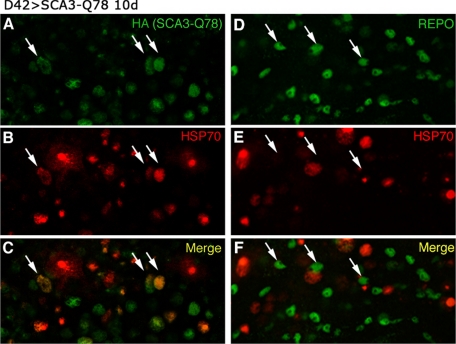
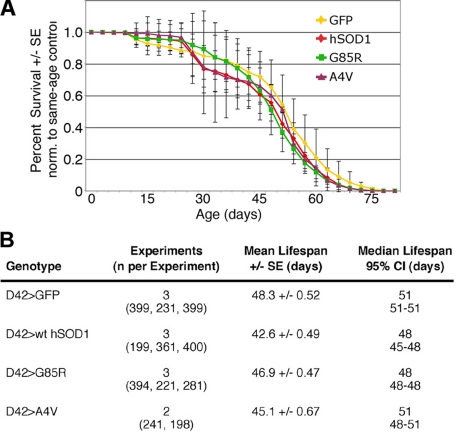
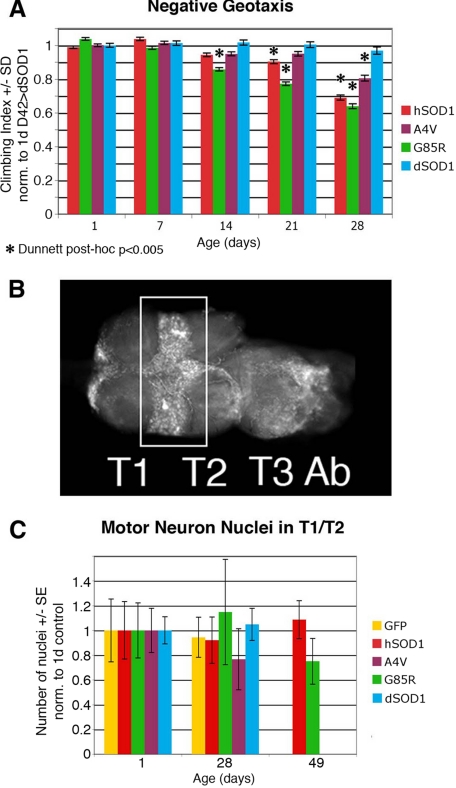
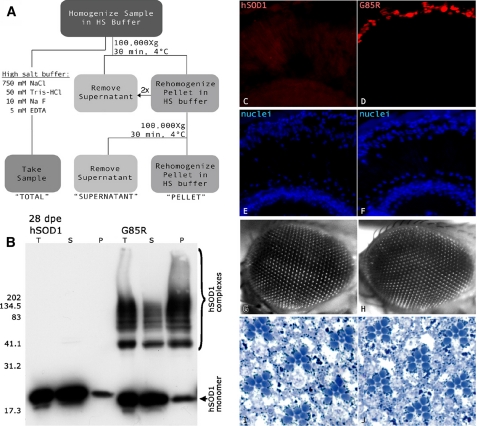
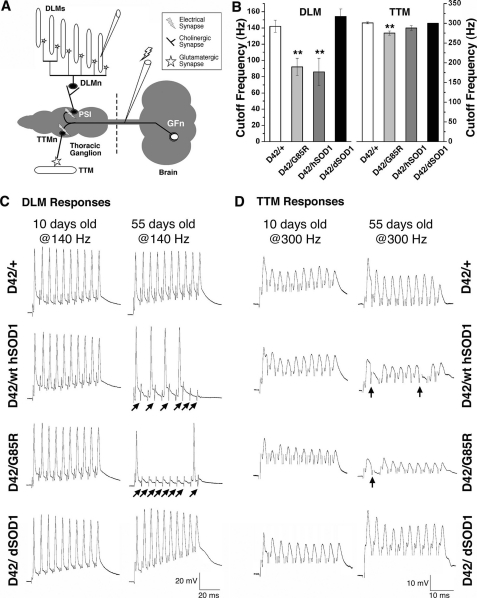
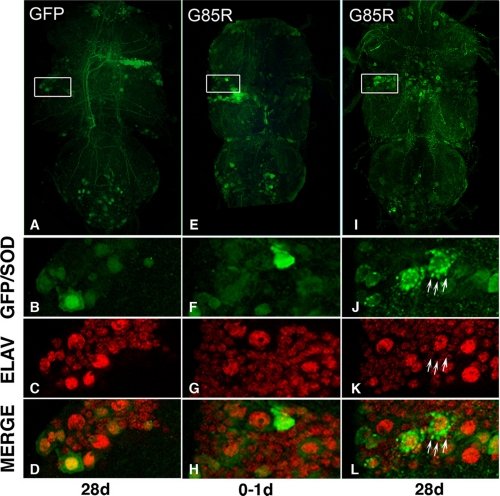
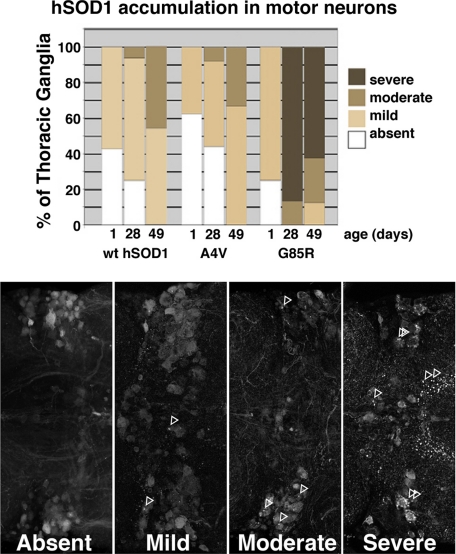
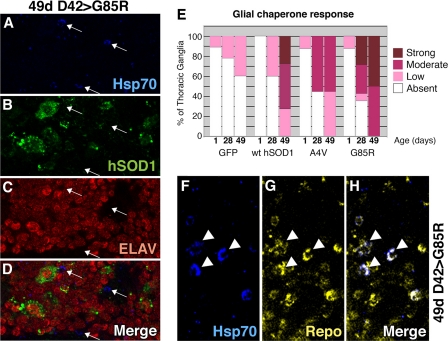
Amyotrophic lateral sclerosis (ALS) is a motor neuron disease that leads to loss of motor function and early death. About 5% of cases are inherited, with the majority of identified linkages in the gene encoding copper, zinc-superoxide dismutase (SOD1). Strong evidence indicates that the SOD1 mutations confer dominant toxicity on the protein. To provide new insight into mechanisms of ALS, we have generated and characterized a model for familial ALS in Drosophila with transgenic expression of human SOD1. Expression of wild type or disease-linked (A4V, G85R) mutants of human SOD1 selectively in motor neurons induced progressive climbing deficits. These effects were accompanied by defective neural circuit electrophysiology, focal accumulation of human SOD1 protein in motor neurons, and a stress response in surrounding glia. However, toxicity was not associated with oligomerization of SOD1 and did not lead to neuronal loss. These studies uncover cell-autonomous injury by SOD1 to motor neurons in vivo, as well as non-autonomous effects on glia, and provide the foundation for new insight into injury and protection of motor neurons in ALS.
肌萎缩侧索硬化症(ALS)是一种运动神经元疾病,会导致运动功能丧失和过早死亡。约5%的病例是遗传性的,大多数已确定的连锁关系存在于编码铜、锌超氧化物歧化酶(SOD1)的基因中。有力证据表明,SOD1突变赋予该蛋白显性毒性。为了深入了解ALS的发病机制,我们构建并鉴定了一种在果蝇中通过转基因表达人SOD1的家族性ALS模型。在运动神经元中选择性表达野生型或疾病相关(A4V、G85R)突变体的人SOD1会导致渐进性攀爬能力缺陷。这些效应伴随着神经回路电生理缺陷、人SOD1蛋白在运动神经元中的局部积累以及周围神经胶质细胞的应激反应。然而,毒性与SOD1的寡聚化无关,也不会导致神经元死亡。这些研究揭示了SOD1在体内对运动神经元的细胞自主损伤以及对神经胶质细胞的非自主效应,为深入了解ALS中运动神经元的损伤和保护机制奠定了基础。