Department of Molecular Biology and Biochemistry, Simon Fraser University, Burnaby, BC, Canada.
Brief Bioinform. 2019 Sep 27;20(5):1685-1698. doi: 10.1093/bib/bby042.
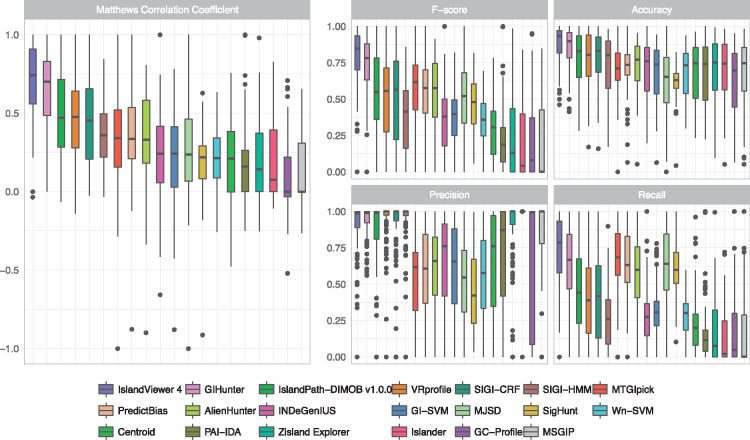
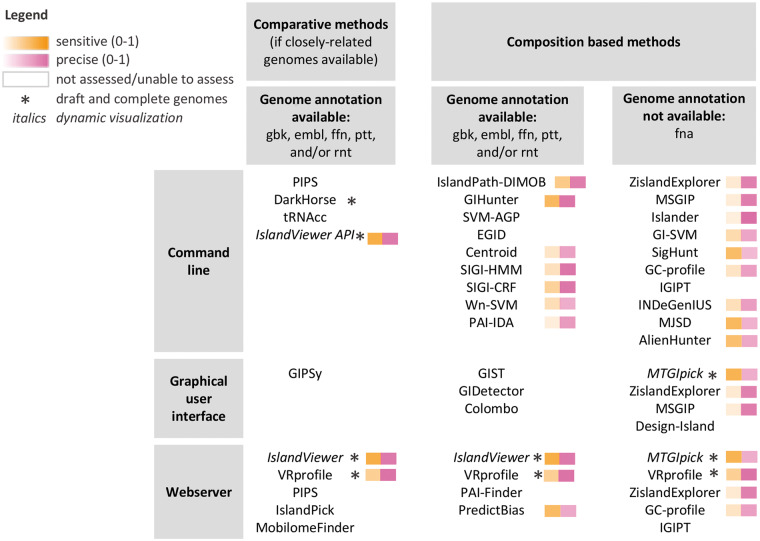
Horizontal gene transfer (also called lateral gene transfer) is a major mechanism for microbial genome evolution, enabling rapid adaptation and survival in specific niches. Genomic islands (GIs), commonly defined as clusters of bacterial or archaeal genes of probable horizontal origin, are of particular medical, environmental and/or industrial interest, as they disproportionately encode virulence factors and some antimicrobial resistance genes and may harbor entire metabolic pathways that confer a specific adaptation (solvent resistance, symbiosis properties, etc). As large-scale analyses of microbial genomes increases, such as for genomic epidemiology investigations of infectious disease outbreaks in public health, there is increased appreciation of the need to accurately predict and track GIs. Over the past decade, numerous computational tools have been developed to tackle the challenges inherent in accurate GI prediction. We review here the main types of GI prediction methods and discuss their advantages and limitations for a routine analysis of microbial genomes in this era of rapid whole-genome sequencing. An assessment is provided of 20 GI prediction software methods that use sequence-composition bias to identify the GIs, using a reference GI data set from 104 genomes obtained using an independent comparative genomics approach. Finally, we present guidelines to assist researchers in effectively identifying these key genomic regions.
水平基因转移(也称为横向基因转移)是微生物基因组进化的主要机制,使微生物能够在特定小生境中快速适应和生存。基因组岛(GI)通常被定义为细菌或古菌基因的簇,其可能具有水平起源,它们具有特别的医学、环境和/或工业意义,因为它们不成比例地编码毒力因子和一些抗生素耐药基因,并且可能包含整个赋予特定适应性(溶剂抗性、共生特性等)的代谢途径。随着对微生物基因组进行大规模分析的增加,例如对公共卫生中传染病爆发的基因组流行病学调查,人们越来越认识到需要准确预测和跟踪 GI。在过去的十年中,已经开发了许多计算工具来解决准确预测 GI 所固有的挑战。我们在这里回顾了主要类型的 GI 预测方法,并讨论了它们在快速全基因组测序时代常规分析微生物基因组的优势和局限性。我们使用来自使用独立比较基因组学方法获得的 104 个基因组的参考 GI 数据集,评估了 20 种使用序列组成偏差来识别 GI 的 GI 预测软件方法。最后,我们提出了指导方针,以帮助研究人员有效地识别这些关键基因组区域。