Milne Iain, Lindner Dominik, Bayer Micha, Husmeier Dirk, McGuire Gráinne, Marshall David F, Wright Frank
Scottish Crop Research Institute, Invergowrie, Dundee, UK.
Bioinformatics. 2009 Jan 1;25(1):126-7. doi: 10.1093/bioinformatics/btn575. Epub 2008 Nov 4.
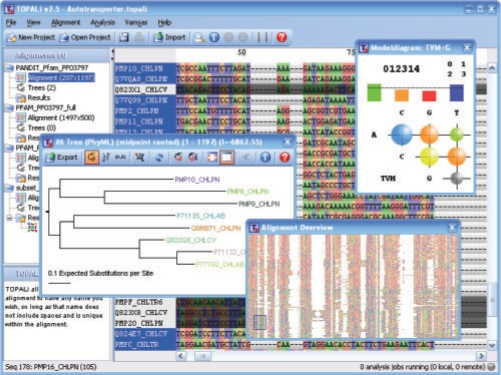
TOPALi v2 simplifies and automates the use of several methods for the evolutionary analysis of multiple sequence alignments. Jobs are submitted from a Java graphical user interface as TOPALi web services to either run remotely on high-performance computing clusters or locally (with multiple cores supported). Methods available include model selection and phylogenetic tree estimation using the Bayesian inference and maximum likelihood (ML) approaches, in addition to recombination detection methods. The optimal substitution model can be selected for protein or nucleic acid (standard, or protein-coding using a codon position model) data using accurate statistical criteria derived from ML co-estimation of the tree and the substitution model. Phylogenetic software available includes PhyML, RAxML and MrBayes.
Freely downloadable from http://www.topali.org for Windows, Mac OS X, Linux and Solaris.
TOPALi v2简化并自动化了多种用于多序列比对进化分析方法的使用。任务通过Java图形用户界面作为TOPALi网络服务提交,以便在高性能计算集群上远程运行或在本地运行(支持多核)。可用方法包括使用贝叶斯推理和最大似然(ML)方法进行模型选择和系统发育树估计,以及重组检测方法。可以使用从树和替换模型的ML共同估计得出的准确统计标准,为蛋白质或核酸(标准,或使用密码子位置模型的蛋白质编码)数据选择最佳替换模型。可用的系统发育软件包括PhyML、RAxML和MrBayes。
可从http://www.topali.org免费下载,适用于Windows、Mac OS X、Linux和Solaris。