Yip Kevin Y, Gerstein Mark
Department of Computer Science, Yale University, New Haven, CT 06511, USA.
Bioinformatics. 2009 Jan 15;25(2):243-50. doi: 10.1093/bioinformatics/btn602. Epub 2008 Nov 17.
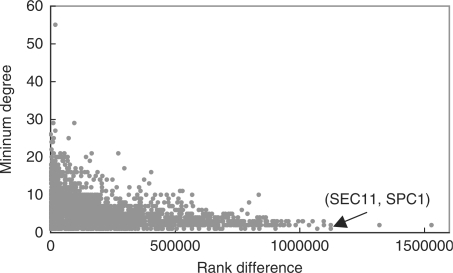
An important problem in systems biology is reconstructing complete networks of interactions between biological objects by extrapolating from a few known interactions as examples. While there are many computational techniques proposed for this network reconstruction task, their accuracy is consistently limited by the small number of high-confidence examples, and the uneven distribution of these examples across the potential interaction space, with some objects having many known interactions and others few.
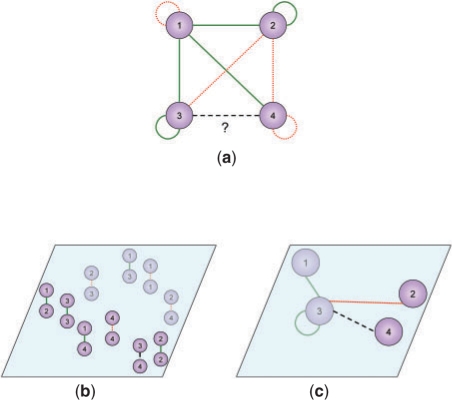
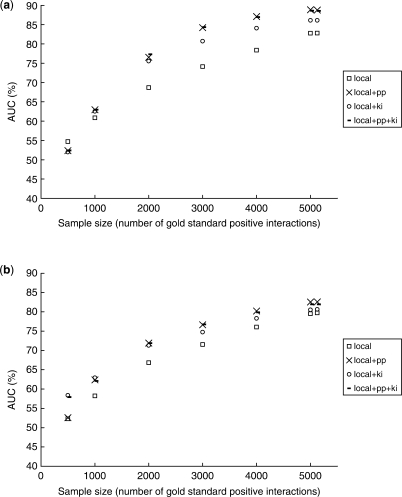
To address this issue, we propose two computational methods based on the concept of training set expansion. They work particularly effectively in conjunction with kernel approaches, which are a popular class of approaches for fusing together many disparate types of features. Both our methods are based on semi-supervised learning and involve augmenting the limited number of gold-standard training instances with carefully chosen and highly confident auxiliary examples. The first method, prediction propagation, propagates highly confident predictions of one local model to another as the auxiliary examples, thus learning from information-rich regions of the training network to help predict the information-poor regions. The second method, kernel initialization, takes the most similar and most dissimilar objects of each object in a global kernel as the auxiliary examples. Using several sets of experimentally verified protein-protein interactions from yeast, we show that training set expansion gives a measurable performance gain over a number of representative, state-of-the-art network reconstruction methods, and it can correctly identify some interactions that are ranked low by other methods due to the lack of training examples of the involved proteins.
系统生物学中的一个重要问题是通过从少数已知的相互作用示例进行推断,来重建生物对象之间完整的相互作用网络。虽然针对此网络重建任务提出了许多计算技术,但它们的准确性始终受到高可信度示例数量少以及这些示例在潜在相互作用空间中分布不均的限制,一些对象有许多已知相互作用,而另一些对象则很少。
为了解决这个问题,我们提出了两种基于训练集扩展概念的计算方法。它们与核方法结合使用时特别有效,核方法是一类流行的方法,用于融合许多不同类型的特征。我们的两种方法都基于半监督学习,并且涉及用精心选择且高度可信的辅助示例来扩充有限数量的金标准训练实例。第一种方法是预测传播,将一个局部模型的高可信度预测作为辅助示例传播到另一个局部模型,从而从训练网络中信息丰富的区域学习,以帮助预测信息贫乏的区域。第二种方法是核初始化,将全局核中每个对象最相似和最不相似的对象作为辅助示例。使用来自酵母的几组经过实验验证的蛋白质 - 蛋白质相互作用,我们表明训练集扩展比许多具有代表性的、当前最先进的网络重建方法在性能上有可测量的提升,并且它可以正确识别一些由于所涉及蛋白质缺乏训练示例而在其他方法中排名较低的相互作用。