Miura Fumihito, Kawaguchi Noriko, Yoshida Mikio, Uematsu Chihiro, Kito Keiji, Sakaki Yoshiyuki, Ito Takashi
Department of Computational Biology, Graduate School of Frontier Sciences, University of Tokyo, Kashiwa 277-8561, Japan.
BMC Genomics. 2008 Nov 29;9:574. doi: 10.1186/1471-2164-9-574.
An ideal format to describe transcriptome would be its composition measured on the scale of absolute numbers of individual mRNAs per cell. It would help not only to precisely grasp the structure of the transcriptome but also to accelerate data exchange and integration.
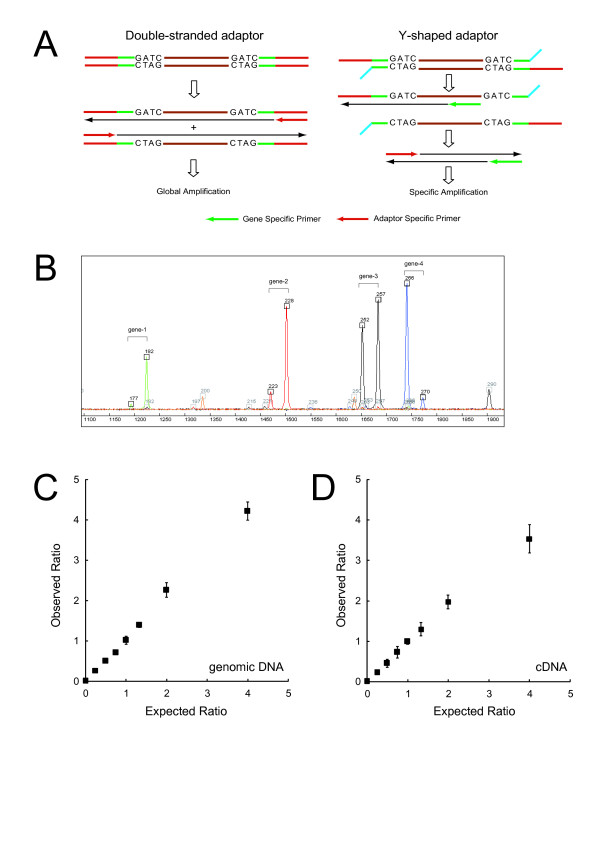
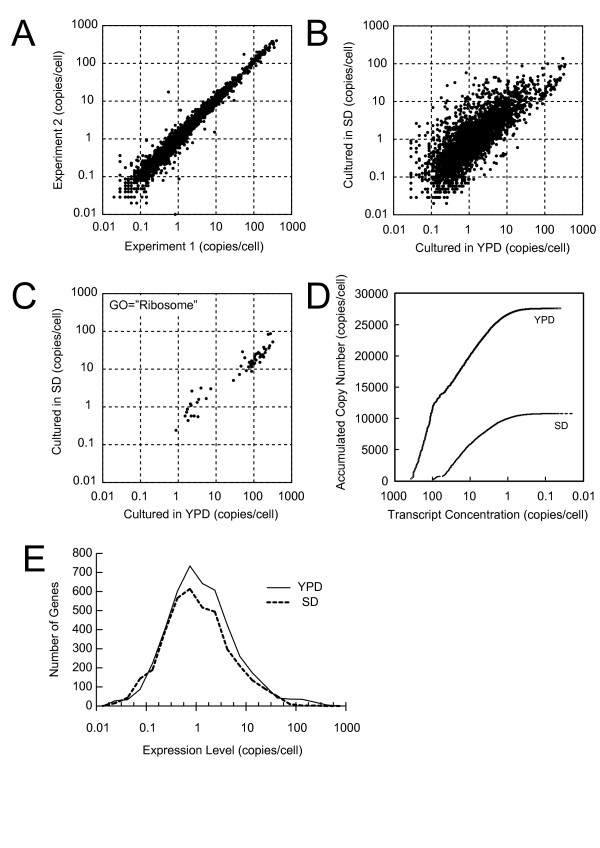
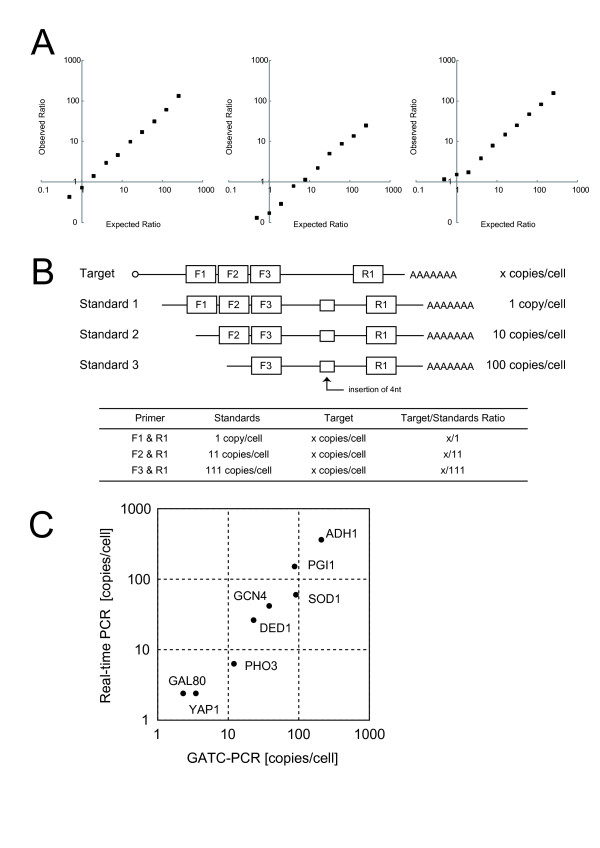
We conceived an idea of competitive PCR between genomic DNA and cDNA. Since the former contains every gene exactly at the same copy number, it can serve as an ideal normalization standard for the latter to obtain stoichiometric composition data of the transcriptome. This data can then be easily converted to absolute quantification data provided with an appropriate calibration. To implement this idea, we improved adaptor-tagged competitive PCR, originally developed for relative quantification of the 3'-end restriction fragment of each cDNA, such that it can be applied to any restriction fragment. We demonstrated that this "generalized" adaptor-tagged competitive PCR (GATC-PCR) can be performed between genomic DNA and cDNA to accurately measure absolute expression level of each mRNA in the budding yeast Saccharomyces cerevisiae. Furthermore, we constructed a large-scale GATC-PCR system to measure absolute expression levels of 5,038 genes to show that the yeast contains more than 30,000 copies of mRNA molecules per cell.
We developed a GATC-PCR method to accurately measure absolute expression levels of mRNAs by means of competitive amplification of genomic and cDNA copies of each gene. A large-scale application of GATC-PCR to the budding yeast transcriptome revealed that it is twice or more as large as previously estimated. This method is flexibly applicable to both targeted and genome-wide analyses of absolute expression levels of mRNAs.
描述转录组的理想形式是在每个细胞中单个mRNA绝对数量的尺度上测量其组成。这不仅有助于精确掌握转录组的结构,还能加速数据交换和整合。
我们构思了一种基因组DNA与cDNA之间竞争性PCR的方法。由于前者精确包含每个基因的相同拷贝数,它可以作为后者的理想标准化标准,以获得转录组的化学计量组成数据。然后,在适当校准的情况下,这些数据可以轻松转换为绝对定量数据。为实现这一想法,我们改进了最初为每个cDNA的3'端限制性片段的相对定量而开发的接头标记竞争性PCR,使其可应用于任何限制性片段。我们证明,这种“通用化”的接头标记竞争性PCR(GATC-PCR)可以在基因组DNA和cDNA之间进行,以准确测量酿酒酵母中每个mRNA的绝对表达水平。此外,我们构建了一个大规模的GATC-PCR系统来测量5038个基因的绝对表达水平,结果表明酵母每个细胞中含有超过30000个mRNA分子拷贝。
我们开发了一种GATC-PCR方法,通过竞争性扩增每个基因的基因组和cDNA拷贝来准确测量mRNA的绝对表达水平。GATC-PCR在酿酒酵母转录组中的大规模应用表明,其规模是先前估计的两倍或更多。该方法可灵活应用于mRNA绝对表达水平的靶向分析和全基因组分析。