Osada Naoki, Innan Hideki
National Institute of Biomedical Innovation, Osaka, Japan.
PLoS Genet. 2008 Dec;4(12):e1000305. doi: 10.1371/journal.pgen.1000305. Epub 2008 Dec 12.
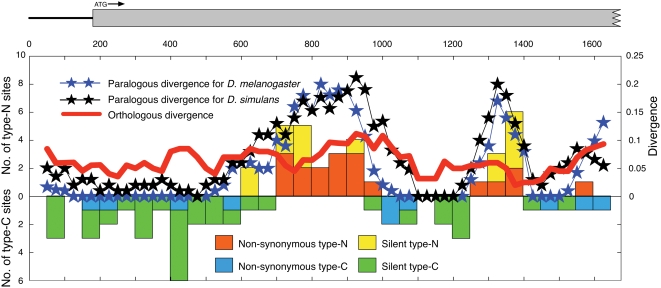
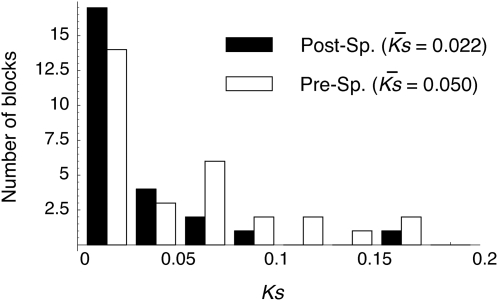
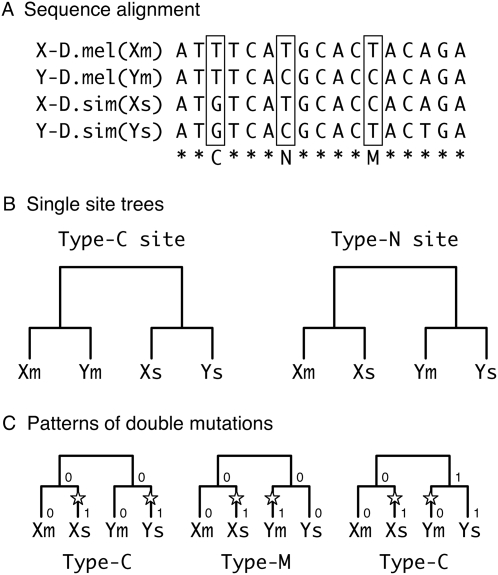
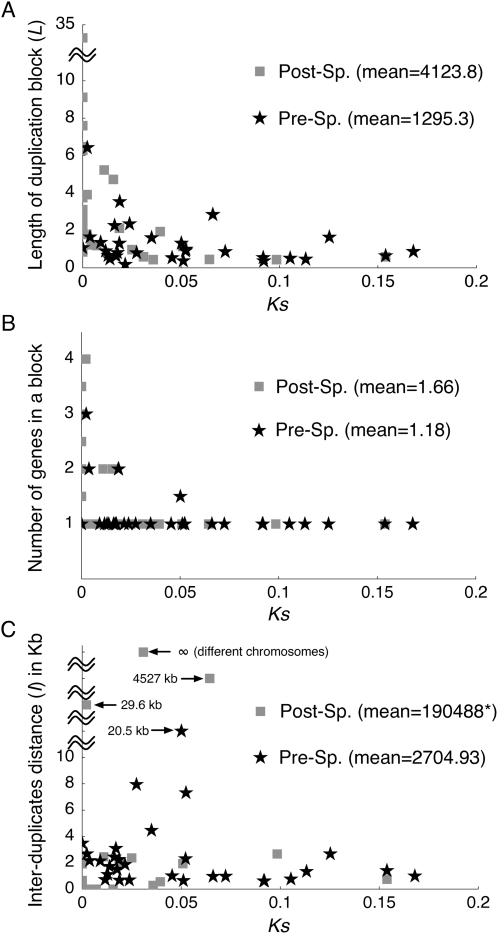
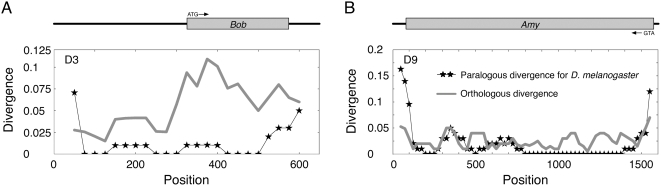
Using the genomic sequences of Drosophila melanogaster subgroup, the pattern of gene duplications was investigated with special attention to interlocus gene conversion. Our fine-scale analysis with careful visual inspections enabled accurate identification of a number of duplicated blocks (genomic regions). The orthologous parts of those duplicated blocks were also identified in the D. simulans and D. sechellia genomes, by which we were able to clearly classify the duplicated blocks into post- and pre-speciation blocks. We found 31 post-speciation duplicated genes, from which the rate of gene duplication (from one copy to two copies) is estimated to be 1.0 x 10(-9) per single-copy gene per year. The role of interlocus gene conversion was observed in several respects in our data: (1) synonymous divergence between a duplicated pair is overall very low. Consequently, the gene duplication rate would be seriously overestimated by counting duplicated genes with low divergence; (2) the sizes of young duplicated blocks are generally large. We postulate that the degeneration of gene conversion around the edges could explain the shrinkage of "identifiable" duplicated regions; and (3) elevated paralogous divergence is observed around the edges in many duplicated blocks, supporting our gene conversion-degeneration model. Our analysis demonstrated that gene conversion between duplicated regions is a common and genome-wide phenomenon in the Drosophila genomes, and that its role should be especially significant in the early stages of duplicated genes. Based on a population genetic prediction, we applied a new genome-scan method to test for signatures of selection for neofunctionalization and found a strong signature in a pair of transporter genes.
利用黑腹果蝇亚组的基因组序列,研究了基因重复模式,并特别关注基因座间基因转换。我们通过仔细的目视检查进行的精细分析,能够准确识别出许多重复块(基因组区域)。在拟果蝇和塞舌尔果蝇的基因组中也鉴定出了这些重复块的直系同源部分,据此我们能够将重复块明确分为物种形成后和物种形成前的块。我们发现了31个物种形成后的重复基因,估计单拷贝基因每年的基因重复率(从一个拷贝到两个拷贝)为1.0×10⁻⁹。我们的数据在几个方面观察到了基因座间基因转换的作用:(1)重复对之间的同义差异总体非常低。因此,通过计算差异低的重复基因来估计基因重复率会严重高估;(2)年轻重复块的大小通常较大。我们推测边缘周围基因转换的退化可以解释“可识别”重复区域的缩小;(3)在许多重复块的边缘观察到旁系同源差异升高,支持我们的基因转换退化模型。我们的分析表明,重复区域之间的基因转换是果蝇基因组中一种普遍且全基因组的现象,其作用在重复基因的早期阶段应该尤为显著。基于群体遗传预测,我们应用了一种新的全基因组扫描方法来测试新功能化选择的特征,在一对转运蛋白基因中发现了强烈的特征。