Gatti Daniel M, Shabalin Andrey A, Lam Tieu-Chong, Wright Fred A, Rusyn Ivan, Nobel Andrew B
Department of Environmental Sciences and Engineering, University of North Carolina, Chapel Hill, North Carolina 27599, USA.
Bioinformatics. 2009 Feb 15;25(4):482-9. doi: 10.1093/bioinformatics/btn648. Epub 2008 Dec 17.
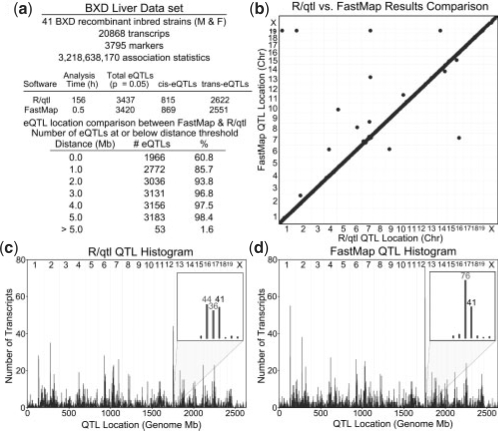
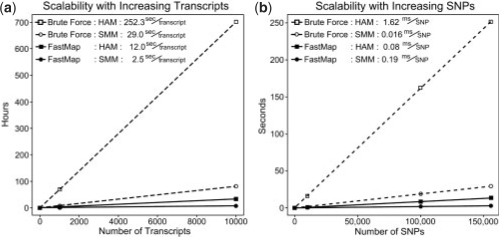
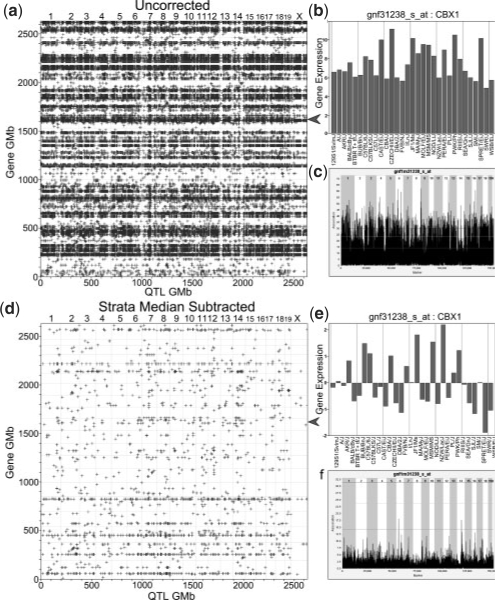
Gene expression Quantitative Trait Locus (eQTL) mapping measures the association between transcript expression and genotype in order to find genomic locations likely to regulate transcript expression. The availability of both gene expression and high-density genotype data has improved our ability to perform eQTL mapping in inbred mouse and other homozygous populations. However, existing eQTL mapping software does not scale well when the number of transcripts and markers are on the order of 10(5) and 10(5)-10(6), respectively.
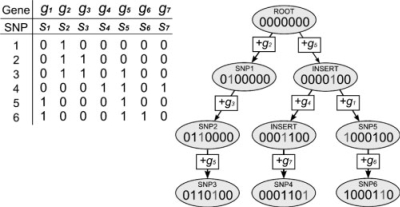
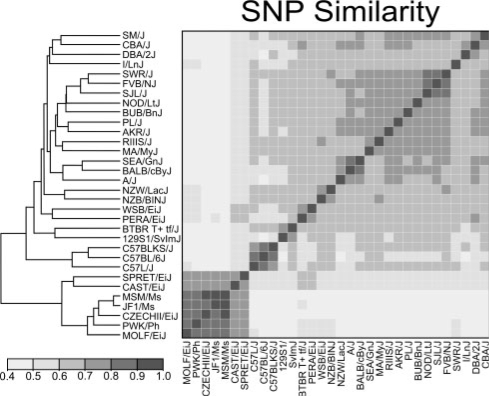
We propose a new method, FastMap, for fast and efficient eQTL mapping in homozygous inbred populations with binary allele calls. FastMap exploits the discrete nature and structure of the measured single nucleotide polymorphisms (SNPs). In particular, SNPs are organized into a Hamming distance-based tree that minimizes the number of arithmetic operations required to calculate the association of a SNP by making use of the association of its parent SNP in the tree. FastMap's tree can be used to perform both single marker mapping and haplotype association mapping over an m-SNP window. These performance enhancements also permit permutation-based significance testing.
The FastMap program and source code are available at the website: http://cebc.unc.edu/fastmap86.html.
基因表达定量性状位点(eQTL)定位通过测量转录本表达与基因型之间的关联,来寻找可能调控转录本表达的基因组位置。基因表达数据和高密度基因型数据的可得性,提高了我们在近交系小鼠和其他纯合群体中进行eQTL定位的能力。然而,当转录本数量和标记数量分别达到10^5和10^5 - 10^6量级时,现有的eQTL定位软件扩展性不佳。
我们提出了一种新方法FastMap,用于在具有二等位基因分型的纯合近交群体中快速高效地进行eQTL定位。FastMap利用了所测单核苷酸多态性(SNP)的离散性质和结构。具体而言,SNP被组织成基于汉明距离的树,通过利用其在树中父SNP的关联,将计算SNP关联所需的算术运算次数减到最少。FastMap的树可用于在m个SNP窗口上进行单标记定位和单倍型关联定位。这些性能提升也允许基于置换的显著性检验。
FastMap程序和源代码可在网站http://cebc.unc.edu/fastmap86.html获取。