MOE Key Laboratory of Bioinformatics and Bioinformatics Division, BNRIST, Department of Automation, Tsinghua University, Beijing, 100084, China.
School of Life Sciences, Center for Synthetic and Systems Biology, Tsinghua University, Beijing, 100084, China.
BMC Bioinformatics. 2020 May 11;21(1):184. doi: 10.1186/s12859-020-3534-6.
With the rapid development of single-cell genomics, technologies for parallel sequencing of the transcriptome and genome in each single cell is being explored in several labs and is becoming available. This brings us the opportunity to uncover association between genotypes and gene expression phenotypes at single-cell level by eQTL analysis on single-cell data. New method is needed for such tasks due to special characteristics of single-cell sequencing data.
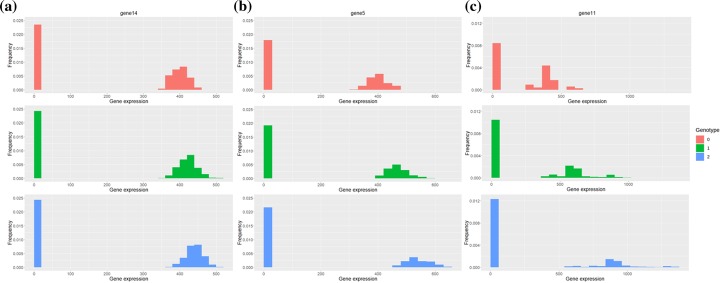
We developed an R package SCeQTL that uses zero-inflated negative binomial regression to do eQTL analysis on single-cell data. It can distinguish two type of gene-expression differences among different genotype groups. It can also be used for finding gene expression variations associated with other grouping factors like cell lineages or cell types.
The SCeQTL method is capable for eQTL analysis on single-cell data as well as detecting associations of gene expression with other grouping factors. The R package of the method is available at https://github.com/XuegongLab/SCeQTL/.
随着单细胞基因组学的快速发展,多个实验室正在探索在单细胞中平行测序转录组和基因组的技术,并且这些技术正在变得可行。这为我们提供了通过单细胞数据的 eQTL 分析在单细胞水平上揭示基因型与基因表达表型之间的关联的机会。由于单细胞测序数据的特殊特征,此类任务需要新的方法。
我们开发了一个 R 包 SCeQTL,该包使用零膨胀负二项式回归在单细胞数据上进行 eQTL 分析。它可以区分不同基因型组之间两种类型的基因表达差异。它还可用于寻找与其他分组因素(如细胞谱系或细胞类型)相关的基因表达变化。
SCeQTL 方法能够对单细胞数据进行 eQTL 分析,并且能够检测基因表达与其他分组因素的关联。该方法的 R 包可在 https://github.com/XuegongLab/SCeQTL/ 上获得。