Sikić Mile, Tomić Sanja, Vlahovicek Kristian
Department of Electronic Systems and Information Processing, Faculty of Electrical Engineering and Computing, University of Zagreb, Zagreb, Croatia.
PLoS Comput Biol. 2009 Jan;5(1):e1000278. doi: 10.1371/journal.pcbi.1000278. Epub 2009 Jan 30.
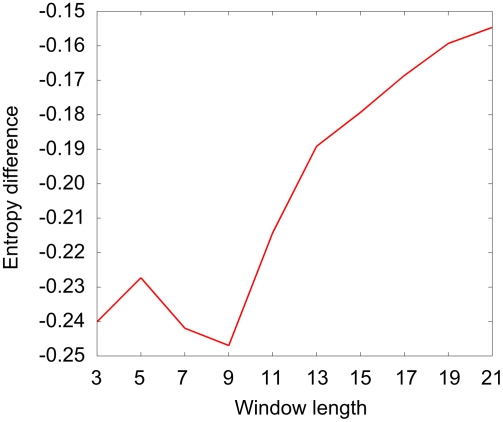
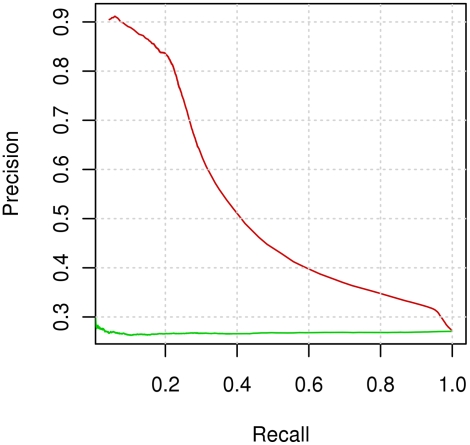
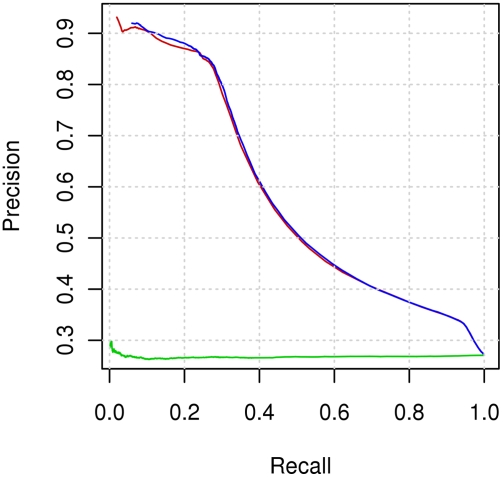
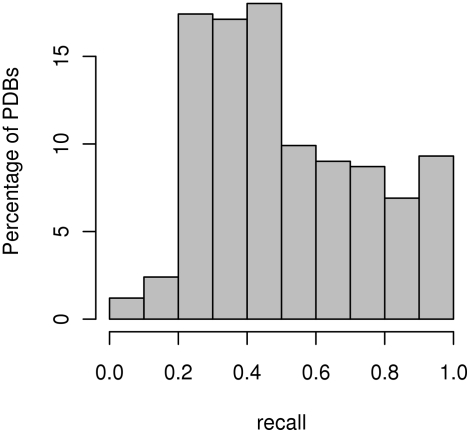
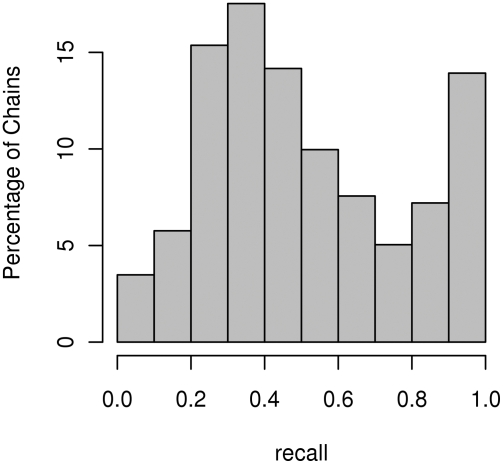
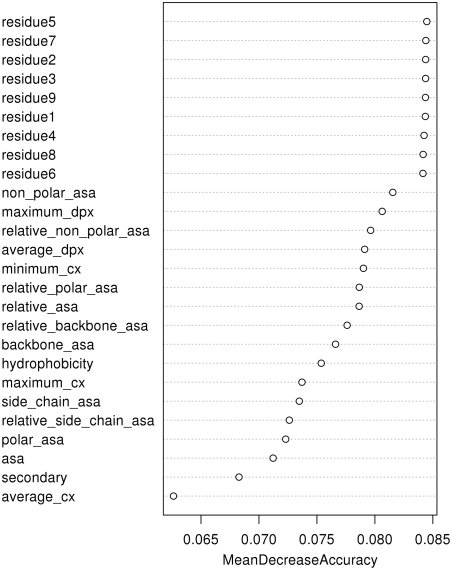
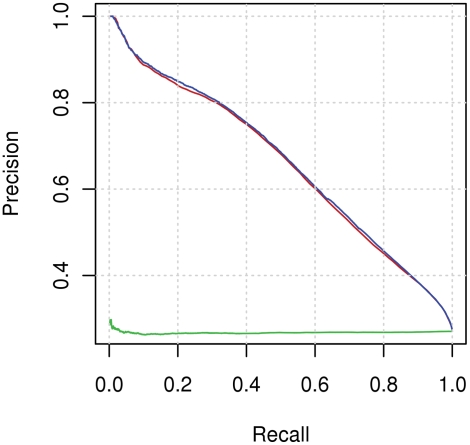
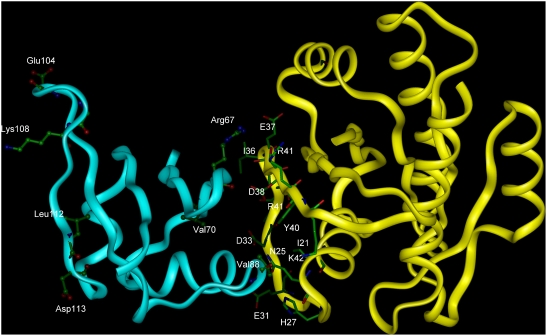
Identifying interaction sites in proteins provides important clues to the function of a protein and is becoming increasingly relevant in topics such as systems biology and drug discovery. Although there are numerous papers on the prediction of interaction sites using information derived from structure, there are only a few case reports on the prediction of interaction residues based solely on protein sequence. Here, a sliding window approach is combined with the Random Forests method to predict protein interaction sites using (i) a combination of sequence- and structure-derived parameters and (ii) sequence information alone. For sequence-based prediction we achieved a precision of 84% with a 26% recall and an F-measure of 40%. When combined with structural information, the prediction performance increases to a precision of 76% and a recall of 38% with an F-measure of 51%. We also present an attempt to rationalize the sliding window size and demonstrate that a nine-residue window is the most suitable for predictor construction. Finally, we demonstrate the applicability of our prediction methods by modeling the Ras-Raf complex using predicted interaction sites as target binding interfaces. Our results suggest that it is possible to predict protein interaction sites with quite a high accuracy using only sequence information.
识别蛋白质中的相互作用位点可为蛋白质功能提供重要线索,并且在系统生物学和药物发现等领域变得越来越重要。尽管有大量关于利用结构衍生信息预测相互作用位点的论文,但仅基于蛋白质序列预测相互作用残基的案例报告却很少。在此,一种滑动窗口方法与随机森林方法相结合,用于预测蛋白质相互作用位点,具体使用(i)基于序列和结构的参数组合,以及(ii)仅使用序列信息。对于基于序列的预测,我们实现了84%的精度、26%的召回率和40%的F值。当与结构信息相结合时,预测性能提高到76%的精度、38%的召回率和51%的F值。我们还尝试对滑动窗口大小进行合理化,并证明九个残基的窗口最适合构建预测器。最后,我们通过将预测的相互作用位点作为目标结合界面来模拟Ras-Raf复合物,证明了我们预测方法的适用性。我们的结果表明,仅使用序列信息就有可能以相当高的准确率预测蛋白质相互作用位点。