Nagamine Nobuyoshi, Shirakawa Takayuki, Minato Yusuke, Torii Kentaro, Kobayashi Hiroki, Imoto Masaya, Sakakibara Yasubumi
Department of Biosciences and Informatics, Keio University, Yokohama, Japan.
PLoS Comput Biol. 2009 Jun;5(6):e1000397. doi: 10.1371/journal.pcbi.1000397. Epub 2009 Jun 5.
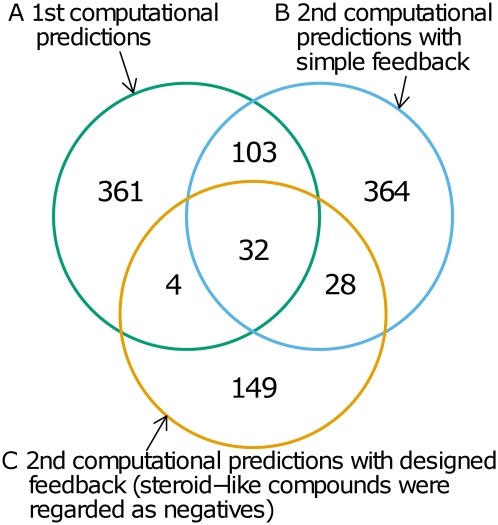
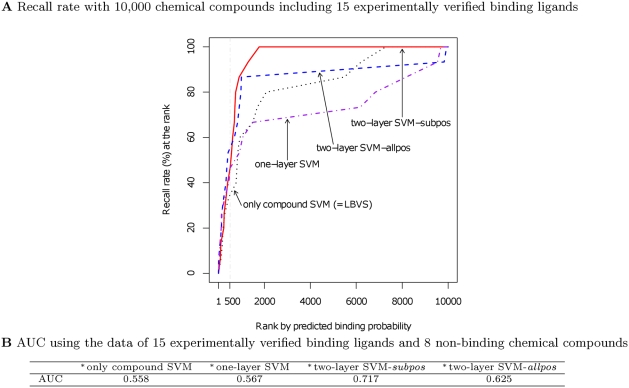
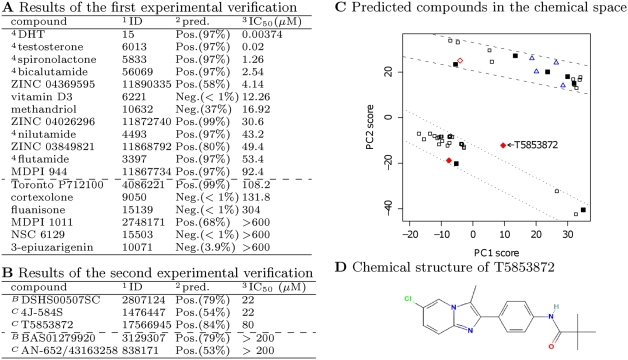
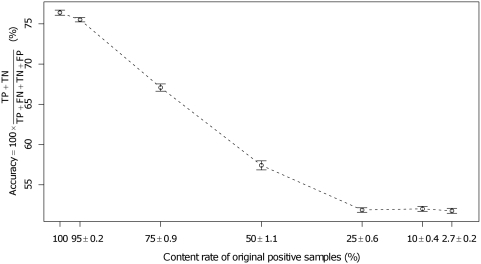
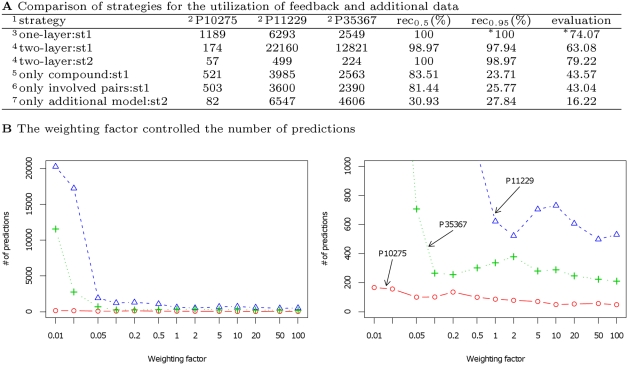
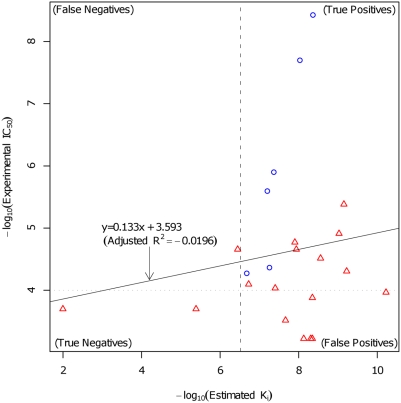
Predictions of interactions between target proteins and potential leads are of great benefit in the drug discovery process. We present a comprehensively applicable statistical prediction method for interactions between any proteins and chemical compounds, which requires only protein sequence data and chemical structure data and utilizes the statistical learning method of support vector machines. In order to realize reasonable comprehensive predictions which can involve many false positives, we propose two approaches for reduction of false positives: (i) efficient use of multiple statistical prediction models in the framework of two-layer SVM and (ii) reasonable design of the negative data to construct statistical prediction models. In two-layer SVM, outputs produced by the first-layer SVM models, which are constructed with different negative samples and reflect different aspects of classifications, are utilized as inputs to the second-layer SVM. In order to design negative data which produce fewer false positive predictions, we iteratively construct SVM models or classification boundaries from positive and tentative negative samples and select additional negative sample candidates according to pre-determined rules. Moreover, in order to fully utilize the advantages of statistical learning methods, we propose a strategy to effectively feedback experimental results to computational predictions with consideration of biological effects of interest. We show the usefulness of our approach in predicting potential ligands binding to human androgen receptors from more than 19 million chemical compounds and verifying these predictions by in vitro binding. Moreover, we utilize this experimental validation as feedback to enhance subsequent computational predictions, and experimentally validate these predictions again. This efficient procedure of the iteration of the in silico prediction and in vitro or in vivo experimental verifications with the sufficient feedback enabled us to identify novel ligand candidates which were distant from known ligands in the chemical space.
预测靶蛋白与潜在先导化合物之间的相互作用在药物发现过程中具有很大的益处。我们提出了一种全面适用的统计预测方法,用于预测任何蛋白质与化合物之间的相互作用,该方法仅需蛋白质序列数据和化学结构数据,并利用支持向量机的统计学习方法。为了实现能够包含许多假阳性结果的合理综合预测,我们提出了两种减少假阳性的方法:(i)在两层支持向量机框架内有效使用多个统计预测模型;(ii)合理设计阴性数据以构建统计预测模型。在两层支持向量机中,由第一层支持向量机模型产生的输出(这些模型由不同的阴性样本构建而成,反映了分类的不同方面)被用作第二层支持向量机的输入。为了设计出产生较少假阳性预测的阴性数据,我们从阳性样本和暂定阴性样本中迭代构建支持向量机模型或分类边界,并根据预先确定的规则选择额外的阴性样本候选物。此外,为了充分利用统计学习方法的优势,我们提出了一种策略,考虑到感兴趣的生物学效应,将实验结果有效地反馈到计算预测中。我们展示了我们的方法在从超过1900万种化合物中预测与人类雄激素受体结合的潜在配体并通过体外结合验证这些预测方面的有用性。此外,我们将这种实验验证作为反馈来增强后续的计算预测,并再次对这些预测进行实验验证。这种通过充分反馈实现计算机模拟预测与体外或体内实验验证迭代的高效程序,使我们能够识别出在化学空间中与已知配体距离较远的新型配体候选物。