Department of Chemistry, University of California - Irvine, 1102 Natural Sciences II, Irvine, California 92697, USA.
J Am Chem Soc. 2009 Mar 4;131(8):2762-3. doi: 10.1021/ja8100825.
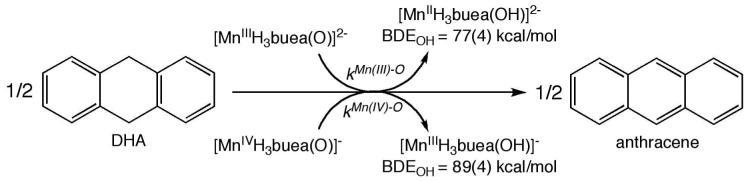
The thermodynamic properties of structurally similar Mn(III) and Mn(IV) complexes have been reinvestigated to understand their reactivity with substrates having C-H bonds. The complexes have the general formula MnH(3)buea(O), where H(3)buea is the tripodal ligand, tris[(N'-tert-butylureaylato)-N-ethylene]aminato. These complexes are unique because of the intramolecular hydrogen-bonding (H-bond) network surrounding the Mn-oxo units. The redox potentials for the Mn(III/IV)(O) couple was incorrectly assigned in earlier reports: the corrected value is -1.0 V vs Cp(2)Fe(+)/Cp(2)Fe in DMSO, while the Mn(IV/V)(O) process is -0.076 under the same conditions. The oxo ligand in the Mn(III)(O) complexes is basic with a pK(a) of 28.3; the basicity of the terminal oxo ligand in the Mn(IV)(O) complex is estimated to be approximately 15. These values were used to re-evalulate the O-H bond dissociation energy (BDE(OH)) of the corresponding Mn(II/III)-OH complexes: BDE(OH) values of 89 and 77 kcal/mol were determined for Mn(III)H(3)buea(OH) and Mn(II)H(3)buea(OH), respectively. Both Mn(O) complexes react with 9,10-dihydroanthracene (DHA) to produce anthracene in nearly quantitative yields. This is surprising based on the low redox potiental of the complexes, suggesting the basicity of the oxo ligand is a major contributor to the observed reactivity. In contrast to the thermodynamic results, a comparative kinetic investigation found that the Mn(III)(O) complex reacts nearly 20 times faster than the Mn(IV)(O) complex. Activation parameters, determined from an Eyring analysis, found that the entropy of activation is significantly different between the two systems (DeltaDeltaS(++) = -35 eu, where DeltaDeltaS(++) = DeltaS(++)(Mn(IV)(O)) - DeltaS(++)(Mn(III)(O)). This unusual kinetic behavior can be explained in the context of the basicity of the oxo ligands that leads to different mechanisms: for Mn(III)H(3)buea(O) a proton transfer-electron transfer mechanism is proposed, whereas for Mn(IV)H(3)buea(O) a hydrogen-atom transfer pathway is likely.
我们重新研究了结构相似的 Mn(III)和 Mn(IV)配合物的热力学性质,以了解它们与具有 C-H 键的底物的反应性。这些配合物具有通式 MnH(3)buea(O),其中 H(3)buea 是三齿配体,三[(N'-叔丁基脲基)-N-乙烯基]氨基。这些配合物因其分子内氢键网络而具有独特性,该网络围绕 Mn-氧单位。早些时候的报告错误地指定了 Mn(III/IV)(O) 对的氧化还原电位:在相同条件下,修正值为-1.0 V 对 Cp(2)Fe(+)/Cp(2)Fe,而 Mn(IV/V)(O) 过程为-0.076。Mn(III)(O) 配合物中的氧配体具有碱性,pK(a)为 28.3;Mn(IV)(O) 配合物中末端氧配体的碱性估计约为 15。这些值用于重新评估相应的 Mn(II/III)-OH 配合物的 O-H 键离解能 (BDE(OH)):分别为 89 和 77 kcal/mol 确定了 Mn(III)H(3)buea(OH)和Mn(II)H(3)buea(OH)的 BDE(OH) 值。两个 Mn(O) 配合物都与 9,10-二氢蒽(DHA)反应,几乎定量生成蒽。这与配合物的低氧化还原电位相比令人惊讶,表明氧配体的碱性是观察到的反应性的主要贡献者。与热力学结果相反,比较动力学研究发现,Mn(III)(O) 配合物的反应速度几乎比 Mn(IV)(O) 配合物快 20 倍。从 Eyring 分析确定的活化参数发现,两个系统之间的活化熵显着不同(DeltaDeltaS(++) = -35 eu,其中 DeltaDeltaS(++) = DeltaS(++)(Mn(IV)(O)) - DeltaS(++)(Mn(III)(O)))。这种不寻常的动力学行为可以在氧配体的碱性的背景下得到解释,这导致了不同的机制:对于 Mn(III)H(3)buea(O),提出了质子转移-电子转移机制,而对于 Mn(IV)H(3)buea(O),可能是氢原子转移途径。