Kadri Sabah, Hinman Veronica, Benos Panayiotis V
Lane Center for Computational Biology, Carnegie Mellon University, Pittsburgh, PA 15213, USA.
BMC Bioinformatics. 2009 Jan 30;10 Suppl 1(Suppl 1):S35. doi: 10.1186/1471-2105-10-S1-S35.
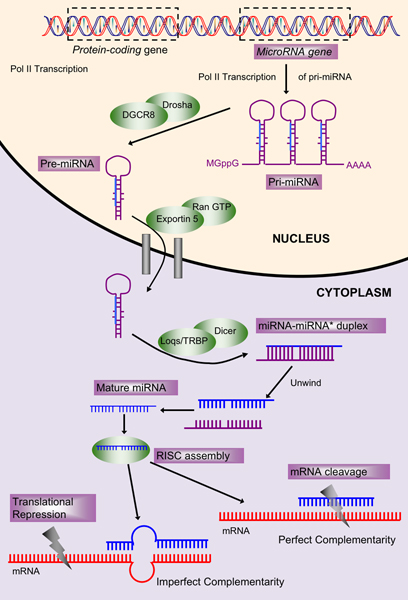
MicroRNAs (miRNAs) are small non-coding single-stranded RNAs (20-23 nts) that are known to act as post-transcriptional and translational regulators of gene expression. Although, they were initially overlooked, their role in many important biological processes, such as development, cell differentiation, and cancer has been established in recent times. In spite of their biological significance, the identification of miRNA genes in newly sequenced organisms is still based, to a large degree, on extensive use of evolutionary conservation, which is not always available.
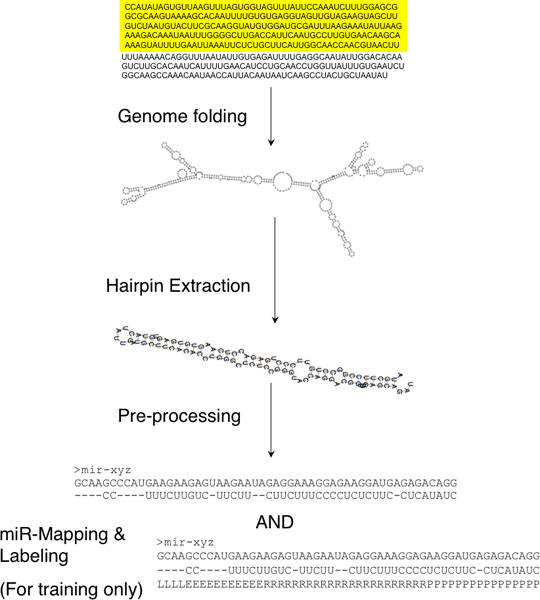
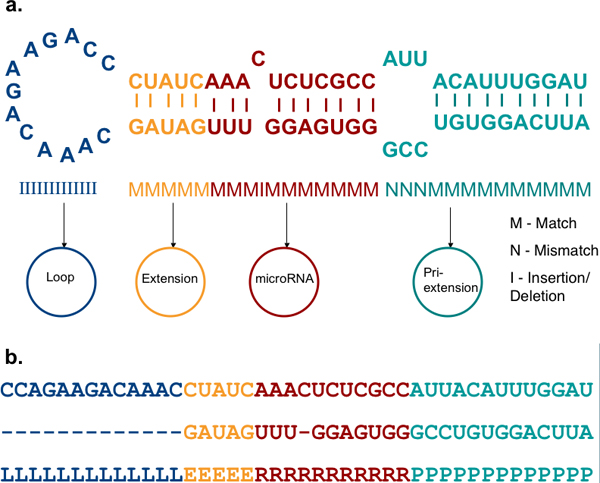
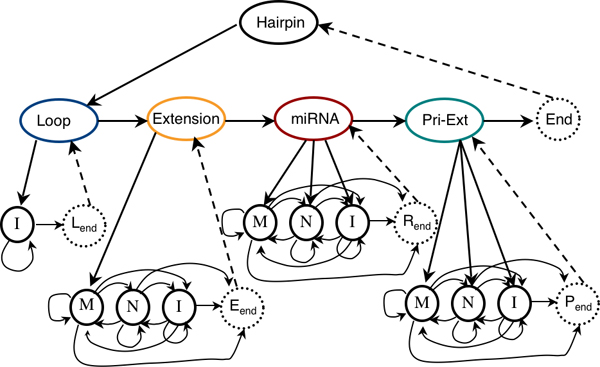
We have developed HHMMiR, a novel approach for de novo miRNA hairpin prediction in the absence of evolutionary conservation. Our method implements a Hierarchical Hidden Markov Model (HHMM) that utilizes region-based structural as well as sequence information of miRNA precursors. We first established a template for the structure of a typical miRNA hairpin by summarizing data from publicly available databases. We then used this template to develop the HHMM topology.
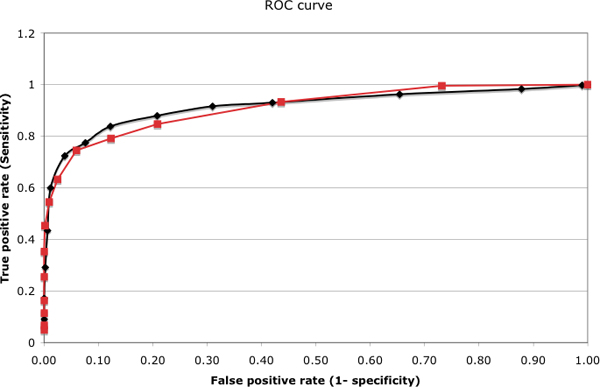
Our algorithm achieved average sensitivity of 84% and specificity of 88%, on 10-fold cross-validation of human miRNA precursor data. We also show that this model, trained on human sequences, works well on hairpins from other vertebrate as well as invertebrate species. Furthermore, the human trained model was able to correctly classify ~97% of plant miRNA precursors. The success of this approach in such a diverse set of species indicates that sequence conservation is not necessary for miRNA prediction. This may lead to efficient prediction of miRNA genes in virtually any organism.
微小RNA(miRNAs)是一类小的非编码单链RNA(20 - 23个核苷酸),已知其作为基因表达的转录后和翻译调节因子发挥作用。尽管它们最初被忽视,但近年来它们在许多重要生物学过程中的作用,如发育、细胞分化和癌症,已被确定。尽管它们具有生物学意义,但在新测序生物中miRNA基因的鉴定在很大程度上仍主要基于对进化保守性的广泛利用,而这种保守性并非总是可用。
我们开发了HHMMiR,这是一种在没有进化保守性的情况下从头预测miRNA发夹结构的新方法。我们的方法实现了一种分层隐马尔可夫模型(HHMM),该模型利用了miRNA前体基于区域的结构以及序列信息。我们首先通过总结来自公开可用数据库的数据,建立了典型miRNA发夹结构的模板。然后我们使用这个模板来开发HHMM拓扑结构。
在对人类miRNA前体数据进行10折交叉验证时,我们的算法平均灵敏度达到84%,特异性达到88%。我们还表明,这个在人类序列上训练的模型,对来自其他脊椎动物以及无脊椎动物物种的发夹结构也能很好地发挥作用。此外,经过人类训练的模型能够正确分类约97%的植物miRNA前体。这种方法在如此多样的物种中的成功表明,miRNA预测并不一定需要序列保守性。这可能会导致几乎在任何生物体中高效预测miRNA基因。