van der Burgt Ate, Fiers Mark W J E, Nap Jan-Peter, van Ham Roeland C H J
Applied Bioinformatics, Plant Research International, Wageningen University & Research Centre, PO Box 16, 6700 AA Wageningen, The Netherlands.
BMC Genomics. 2009 Apr 30;10:204. doi: 10.1186/1471-2164-10-204.
MicroRNAs (miRNAs), short approximately 21-nucleotide RNA molecules, play an important role in post-transcriptional regulation of gene expression. The number of known miRNA hairpins registered in the miRBase database is rapidly increasing, but recent reports suggest that many miRNAs with restricted temporal or tissue-specific expression remain undiscovered. Various strategies for in silico miRNA identification have been proposed to facilitate miRNA discovery. Notably support vector machine (SVM) methods have recently gained popularity. However, a drawback of these methods is that they do not provide insight into the biological properties of miRNA sequences.
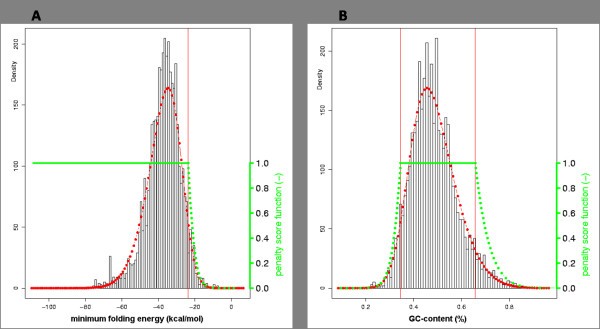
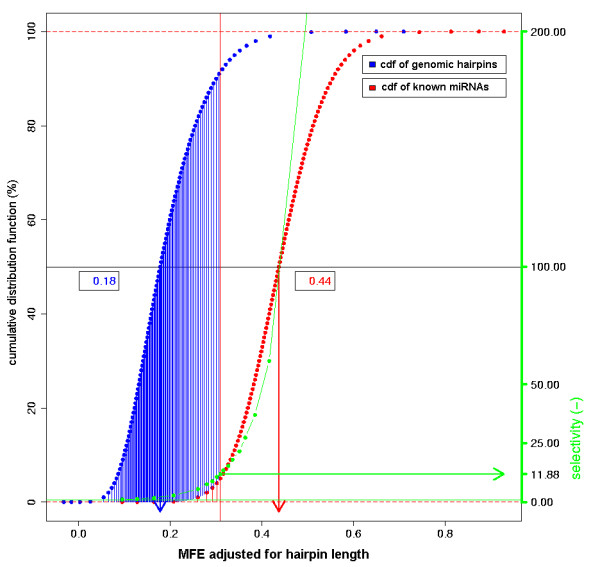
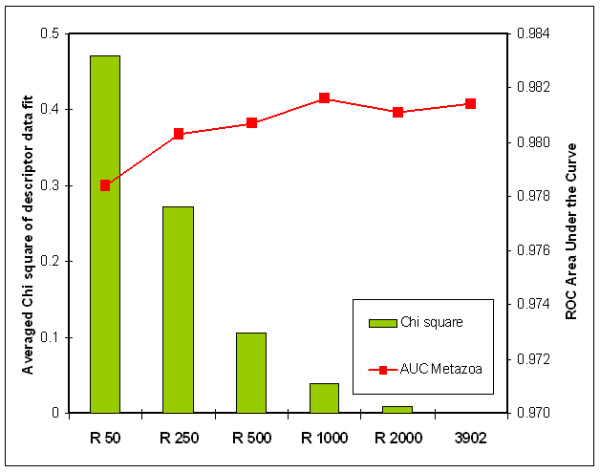
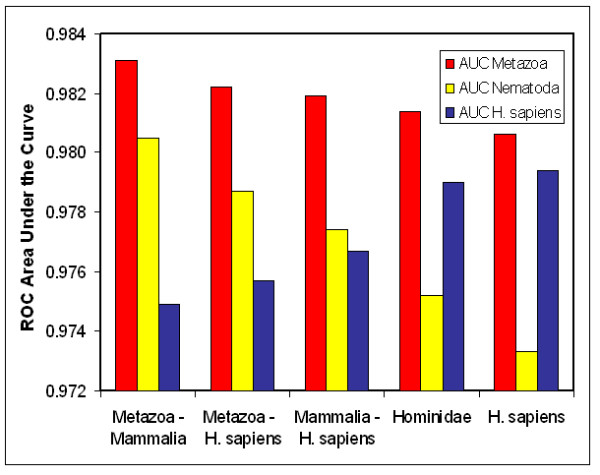
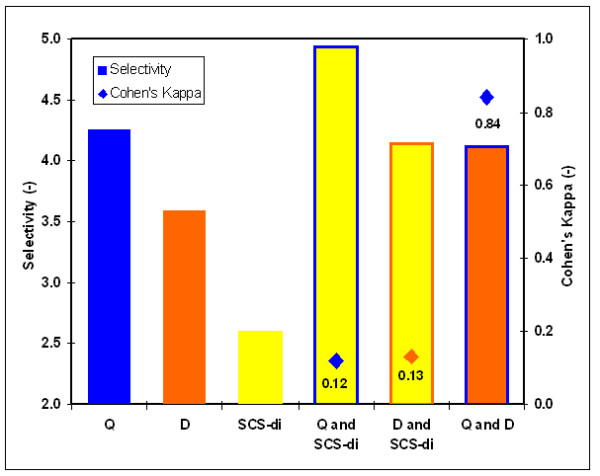
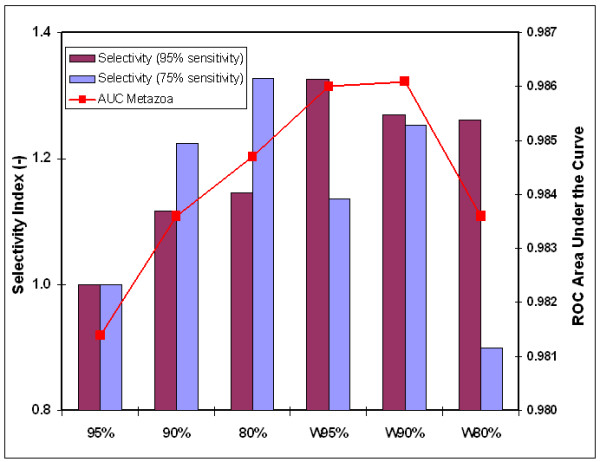
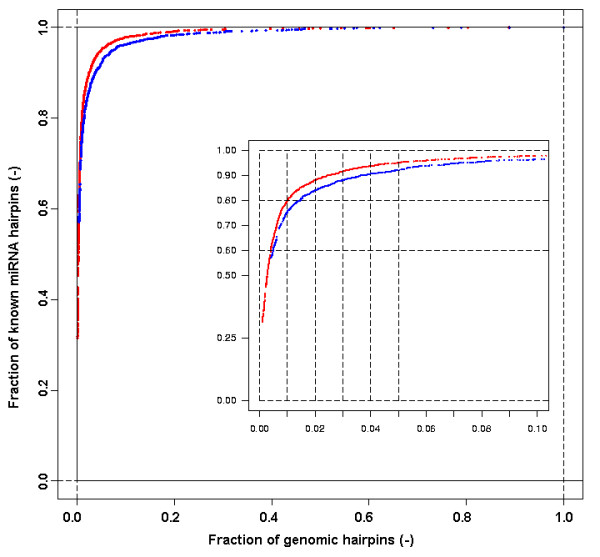
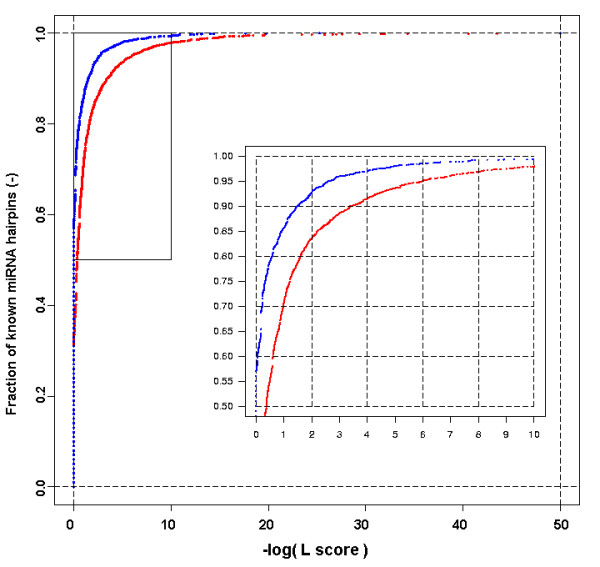
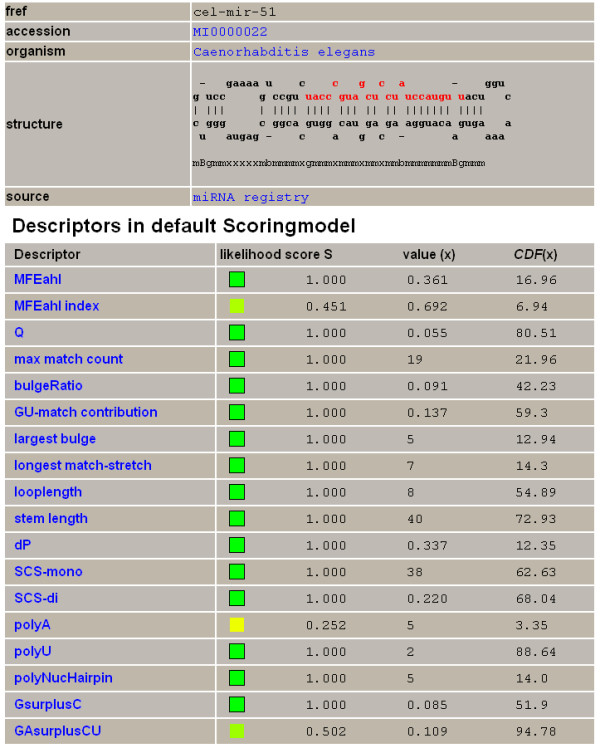
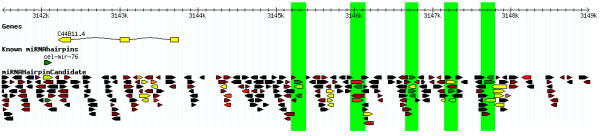
We here propose a new strategy for miRNA hairpin prediction in which the likelihood that a genomic hairpin is a true miRNA hairpin is evaluated based on statistical distributions of observed biological variation of properties (descriptors) of known miRNA hairpins. These distributions are transformed into a single and continuous outcome classifier called the L score. Using a dataset of known miRNA hairpins from the miRBase database and an exhaustive set of genomic hairpins identified in the genome of Caenorhabditis elegans, a subset of 18 most informative descriptors was selected after detailed analysis of correlation among and discriminative power of individual descriptors. We show that the majority of previously identified miRNA hairpins have high L scores, that the method outperforms miRNA prediction by threshold filtering and that it is more transparent than SVM classifiers.
The L score is applicable as a prediction classifier with high sensitivity for novel miRNA hairpins. The L-score approach can be used to rank and select interesting miRNA hairpin candidates for downstream experimental analysis when coupled to a genome-wide set of in silico-identified hairpins or to facilitate the analysis of large sets of putative miRNA hairpin loci obtained in deep-sequencing efforts of small RNAs. Moreover, the in-depth analyses of miRNA hairpins descriptors preceding and determining the L score outcome could be used as an extension to miRBase entries to help increase the reliability and biological relevance of the miRNA registry.
微小RNA(miRNA)是长度约为21个核苷酸的短RNA分子,在基因表达的转录后调控中发挥重要作用。miRBase数据库中注册的已知miRNA发夹数量正在迅速增加,但最近的报告表明,许多具有受限时间或组织特异性表达的miRNA仍未被发现。为促进miRNA的发现,人们提出了各种基于计算机的miRNA识别策略。值得注意的是,支持向量机(SVM)方法最近受到了广泛关注。然而,这些方法的一个缺点是它们无法深入了解miRNA序列的生物学特性。
我们在此提出一种新的miRNA发夹预测策略,即根据已知miRNA发夹的特性(描述符)的观察生物学变异的统计分布,评估基因组发夹是真正miRNA发夹的可能性。这些分布被转换为一个单一的连续结果分类器,称为L分数。使用来自miRBase数据库的已知miRNA发夹数据集以及在秀丽隐杆线虫基因组中鉴定出的一组详尽的基因组发夹,在详细分析各个描述符之间的相关性和判别力后,选择了18个最具信息性的描述符子集。我们表明,大多数先前鉴定的miRNA发夹具有较高的L分数,该方法优于通过阈值过滤进行的miRNA预测,并且比SVM分类器更具透明度。
L分数可作为对新型miRNA发夹具有高灵敏度的预测分类器。当与全基因组范围内基于计算机识别的发夹集结合使用时,L分数方法可用于对有趣的miRNA发夹候选物进行排名和选择,以进行下游实验分析,或促进对在小RNA深度测序工作中获得的大量假定miRNA发夹位点的分析。此外,对决定L分数结果的miRNA发夹描述符的深入分析可作为miRBase条目的扩展,以帮助提高miRNA登记的可靠性和生物学相关性。