Rockman Matthew V, Kruglyak Leonid
Lewis-Sigler Institute for Integrative Genomics, Princeton University, Princeton, New Jersey, United States of America.
PLoS Genet. 2009 Mar;5(3):e1000419. doi: 10.1371/journal.pgen.1000419. Epub 2009 Mar 13.
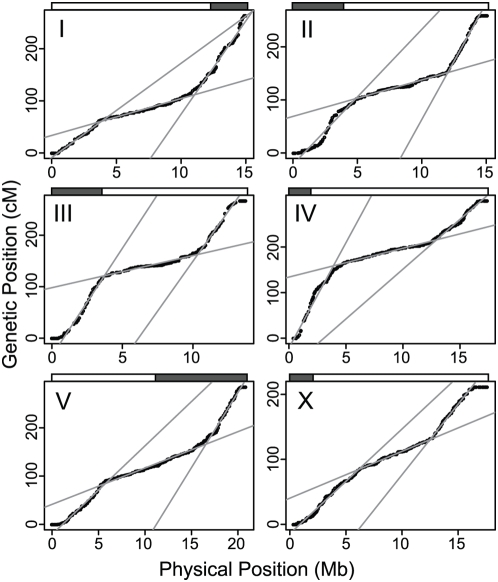
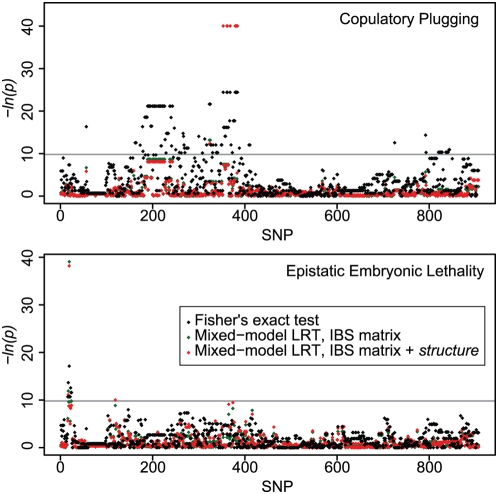
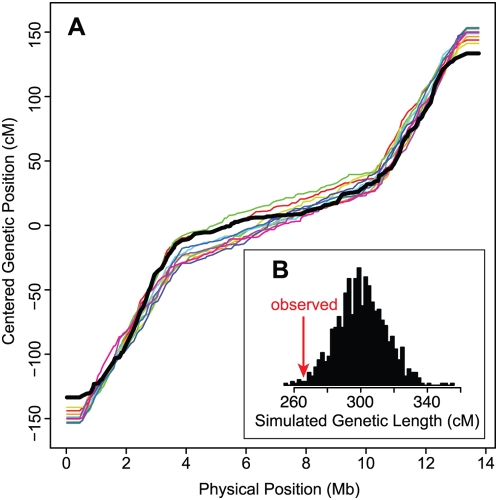
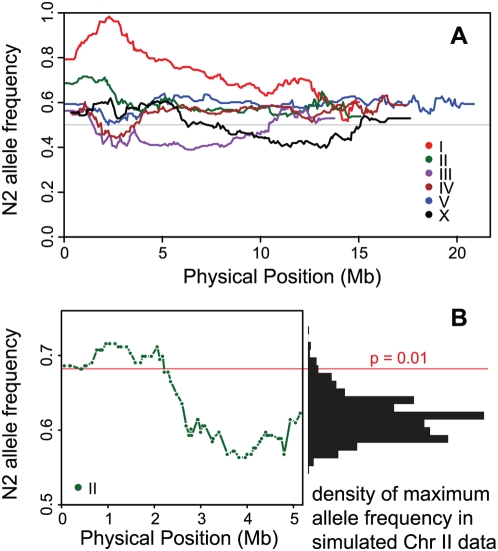
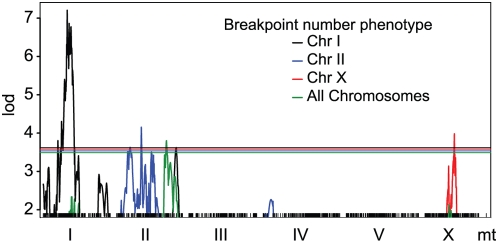
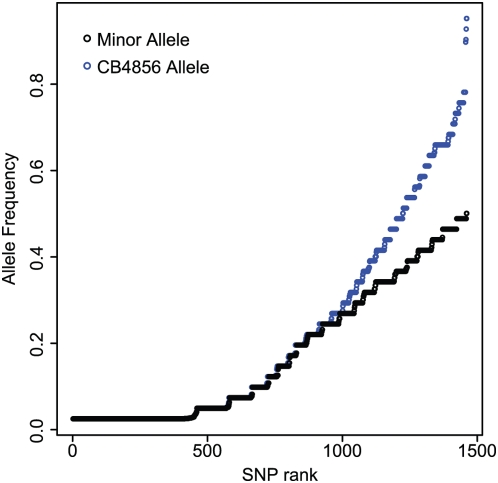
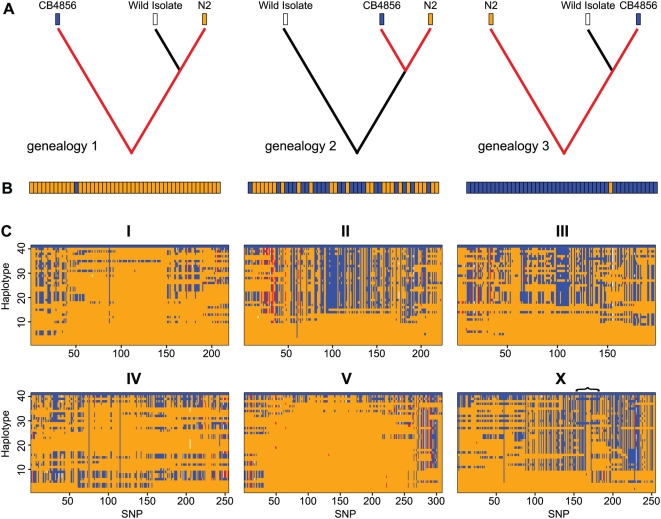
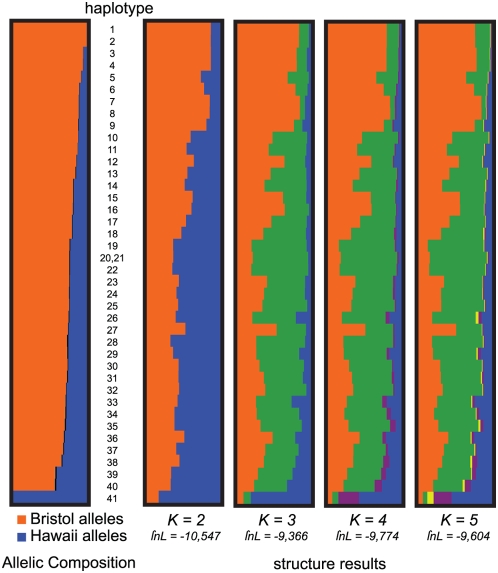
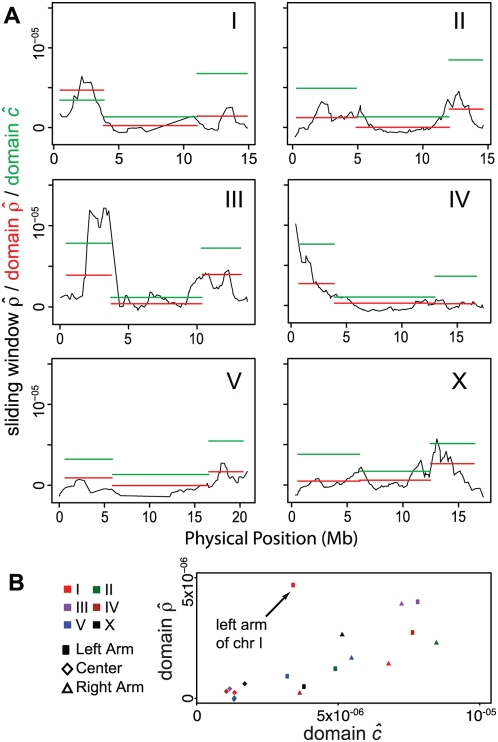
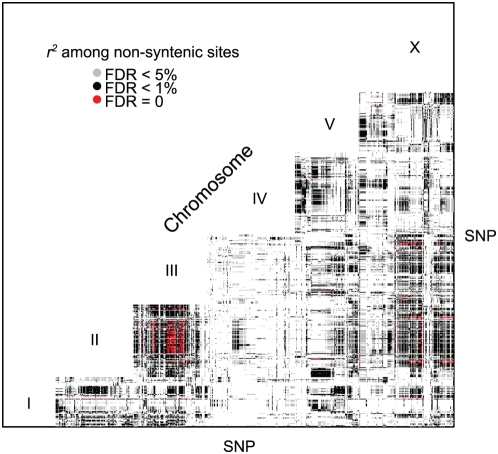
Recombination rate and linkage disequilibrium, the latter a function of population genomic processes, are the critical parameters for mapping by linkage and association, and their patterns in Caenorhabditis elegans are poorly understood. We performed high-density SNP genotyping on a large panel of recombinant inbred advanced intercross lines (RIAILs) of C. elegans to characterize the landscape of recombination and, on a panel of wild strains, to characterize population genomic patterns. We confirmed that C. elegans autosomes exhibit discrete domains of nearly constant recombination rate, and we show, for the first time, that the pattern holds for the X chromosome as well. The terminal domains of each chromosome, spanning about 7% of the genome, exhibit effectively no recombination. The RIAILs exhibit a 5.3-fold expansion of the genetic map. With median marker spacing of 61 kb, they are a powerful resource for mapping quantitative trait loci in C. elegans. Among 125 wild isolates, we identified only 41 distinct haplotypes. The patterns of genotypic similarity suggest that some presumed wild strains are laboratory contaminants. The Hawaiian strain, CB4856, exhibits genetic isolation from the remainder of the global population, whose members exhibit ample evidence of intercrossing and recombining. The population effective recombination rate, estimated from the pattern of linkage disequilibrium, is correlated with the estimated meiotic recombination rate, but its magnitude implies that the effective rate of outcrossing is extremely low, corroborating reports of selection against recombinant genotypes. Despite the low population, effective recombination rate and extensive linkage disequilibrium among chromosomes, which are techniques that account for background levels of genomic similarity, permit association mapping in wild C. elegans strains.
重组率和连锁不平衡(后者是群体基因组过程的一个函数)是通过连锁和关联进行图谱绘制的关键参数,而它们在秀丽隐杆线虫中的模式却鲜为人知。我们对一大组秀丽隐杆线虫的重组近交高级杂交系(RIAILs)进行了高密度单核苷酸多态性(SNP)基因分型,以描绘重组图谱,并且对一组野生菌株进行了基因分型,以描绘群体基因组模式。我们证实秀丽隐杆线虫常染色体呈现出重组率几乎恒定的离散区域,并且首次表明这种模式在X染色体上也成立。每条染色体的末端区域,覆盖约7%的基因组,实际上没有重组现象。RIAILs的遗传图谱扩展了5.3倍。标记的中位数间距为61 kb,它们是用于绘制秀丽隐杆线虫数量性状基因座的强大资源。在125个野生分离株中,我们仅鉴定出41种不同的单倍型。基因型相似性模式表明,一些假定的野生菌株是实验室污染物。夏威夷菌株CB4856与全球其他群体存在遗传隔离,而其他群体成员有充分的杂交和重组证据。根据连锁不平衡模式估计的群体有效重组率与估计的减数分裂重组率相关,但其大小意味着异交的有效率极低,这证实了针对重组基因型选择的报道。尽管群体有效重组率较低且染色体间存在广泛的连锁不平衡,但考虑基因组相似性背景水平的技术仍允许在野生秀丽隐杆线虫菌株中进行关联图谱绘制。