White James Robert, Nagarajan Niranjan, Pop Mihai
Applied Mathematics and Scientific Computation Program, Center for Bioinformatics and Computational Biology, University of Maryland, College Park, Maryland, United States of America.
PLoS Comput Biol. 2009 Apr;5(4):e1000352. doi: 10.1371/journal.pcbi.1000352. Epub 2009 Apr 10.
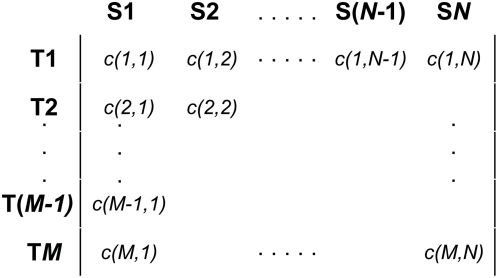
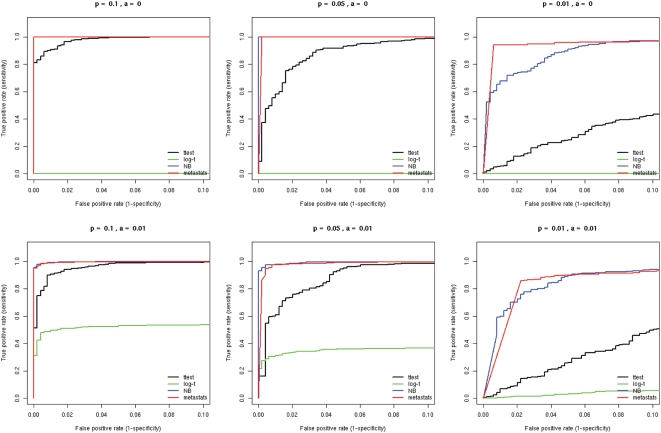
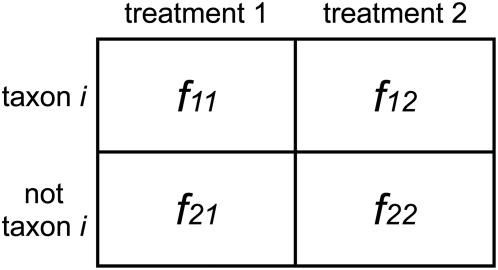
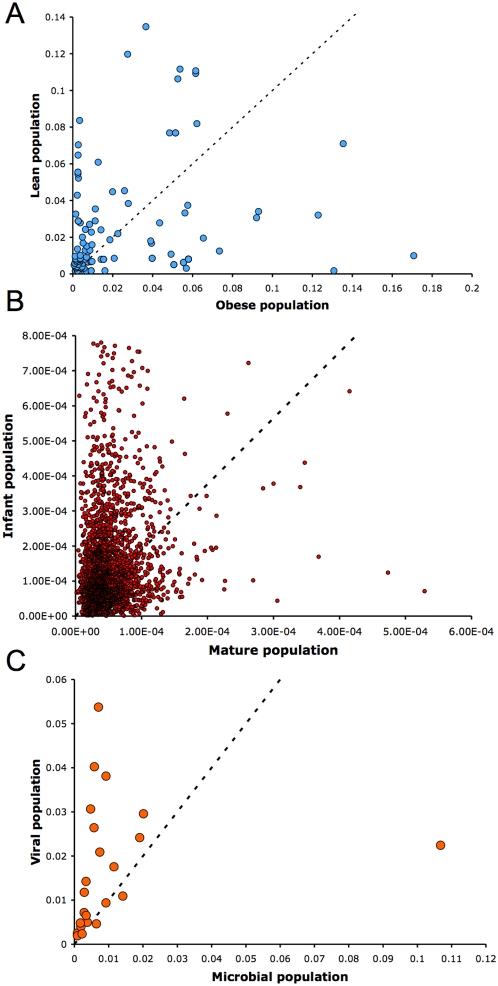
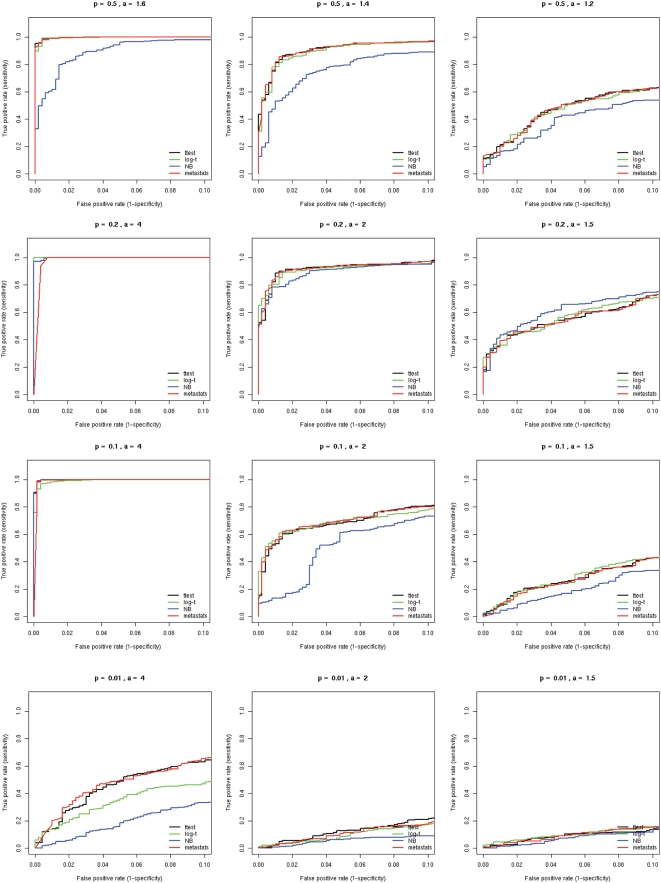
Numerous studies are currently underway to characterize the microbial communities inhabiting our world. These studies aim to dramatically expand our understanding of the microbial biosphere and, more importantly, hope to reveal the secrets of the complex symbiotic relationship between us and our commensal bacterial microflora. An important prerequisite for such discoveries are computational tools that are able to rapidly and accurately compare large datasets generated from complex bacterial communities to identify features that distinguish them.We present a statistical method for comparing clinical metagenomic samples from two treatment populations on the basis of count data (e.g. as obtained through sequencing) to detect differentially abundant features. Our method, Metastats, employs the false discovery rate to improve specificity in high-complexity environments, and separately handles sparsely-sampled features using Fisher's exact test. Under a variety of simulations, we show that Metastats performs well compared to previously used methods, and significantly outperforms other methods for features with sparse counts. We demonstrate the utility of our method on several datasets including a 16S rRNA survey of obese and lean human gut microbiomes, COG functional profiles of infant and mature gut microbiomes, and bacterial and viral metabolic subsystem data inferred from random sequencing of 85 metagenomes. The application of our method to the obesity dataset reveals differences between obese and lean subjects not reported in the original study. For the COG and subsystem datasets, we provide the first statistically rigorous assessment of the differences between these populations. The methods described in this paper are the first to address clinical metagenomic datasets comprising samples from multiple subjects. Our methods are robust across datasets of varied complexity and sampling level. While designed for metagenomic applications, our software can also be applied to digital gene expression studies (e.g. SAGE). A web server implementation of our methods and freely available source code can be found at http://metastats.cbcb.umd.edu/.
目前有许多研究正在进行,以描绘我们这个世界中的微生物群落特征。这些研究旨在大幅拓展我们对微生物生物圈的理解,更重要的是,有望揭示我们与共生细菌微生物群之间复杂共生关系的奥秘。进行此类发现的一个重要前提是要有能够快速且准确地比较从复杂细菌群落生成的大型数据集,以识别区分它们的特征的计算工具。我们提出了一种基于计数数据(例如通过测序获得的数据)来比较来自两个治疗群体的临床宏基因组样本,以检测差异丰富特征的统计方法。我们的方法“Metastats”利用错误发现率来提高在高复杂性环境中的特异性,并使用Fisher精确检验分别处理稀疏采样的特征。在各种模拟中,我们表明与先前使用的方法相比,“Metastats”表现良好,并且在处理计数稀疏的特征时明显优于其他方法。我们在几个数据集上展示了我们方法的实用性,包括肥胖和瘦人的肠道微生物群的16S rRNA调查、婴儿和成熟肠道微生物群的COG功能概况,以及从85个宏基因组的随机测序推断出的细菌和病毒代谢子系统数据。将我们的方法应用于肥胖数据集揭示了原始研究中未报告的肥胖和瘦人受试者之间的差异。对于COG和子系统数据集,我们首次对这些群体之间的差异进行了严格的统计学评估。本文所述的方法是首次针对包含来自多个受试者样本的临床宏基因组数据集。我们的方法在不同复杂性和采样水平的数据集上都很稳健。虽然是为宏基因组应用而设计的,但我们的软件也可应用于数字基因表达研究(例如SAGE)。我们方法的网络服务器实现和免费可用的源代码可在http://metastats.cbcb.umd.edu/找到。