Liu Bo, Pop Mihai
Center for Bioinformatics and Computational Biology, Institute for Advanced Computer Studies, University of Maryland, College Park, MD 20742, USA.
BMC Proc. 2011 May 28;5 Suppl 2(Suppl 2):S9. doi: 10.1186/1753-6561-5-S2-S9.
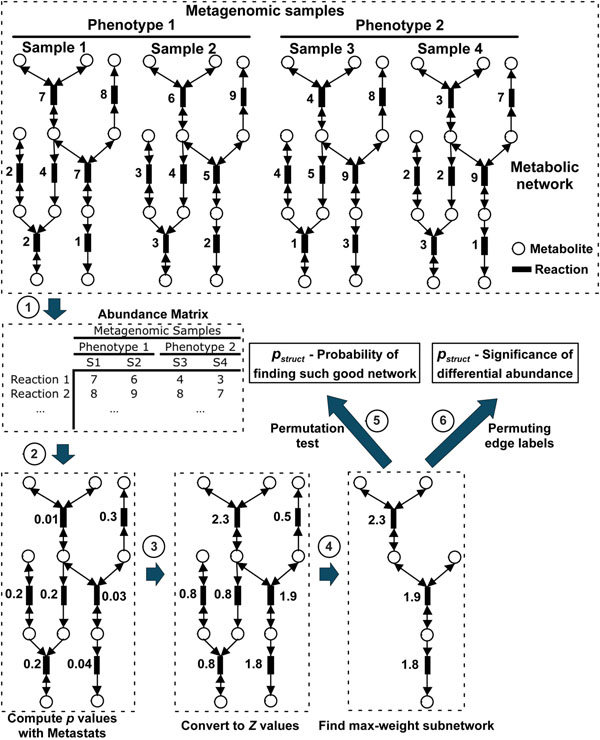
Enabled by rapid advances in sequencing technology, metagenomic studies aim to characterize entire communities of microbes bypassing the need for culturing individual bacterial members. One major goal of metagenomic studies is to identify specific functional adaptations of microbial communities to their habitats. The functional profile and the abundances for a sample can be estimated by mapping metagenomic sequences to the global metabolic network consisting of thousands of molecular reactions. Here we describe a powerful analytical method (MetaPath) that can identify differentially abundant pathways in metagenomic datasets, relying on a combination of metagenomic sequence data and prior metabolic pathway knowledge.
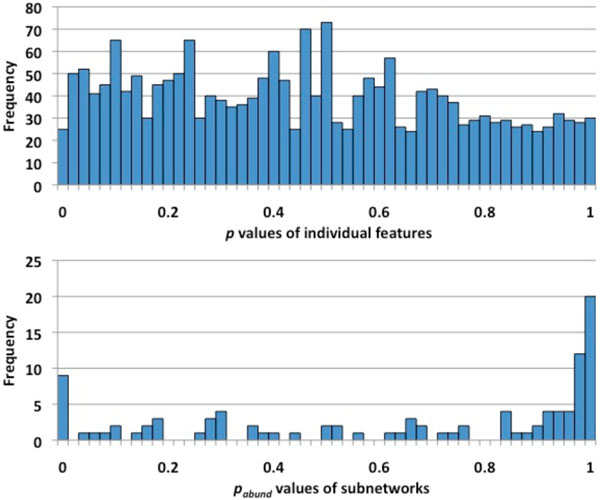
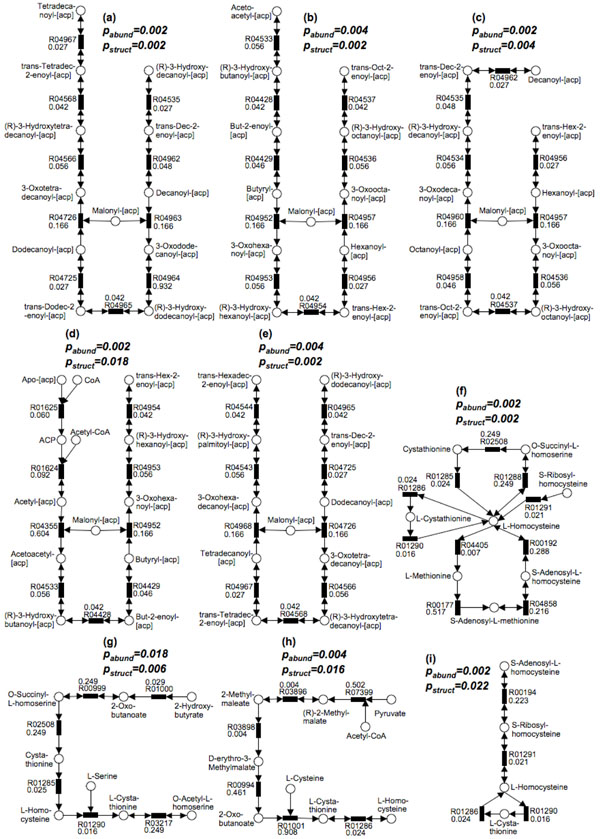
First, we introduce a scoring function for an arbitrary subnetwork and find the max-weight subnetwork in the global network by a greedy search algorithm. Then we compute two p values (pabund and pstruct) using nonparametric approaches to answer two different statistical questions: (1) is this subnetwork differentically abundant? (2) What is the probability of finding such good subnetworks by chance given the data and network structure? Finally, significant metabolic subnetworks are discovered based on these two p values.
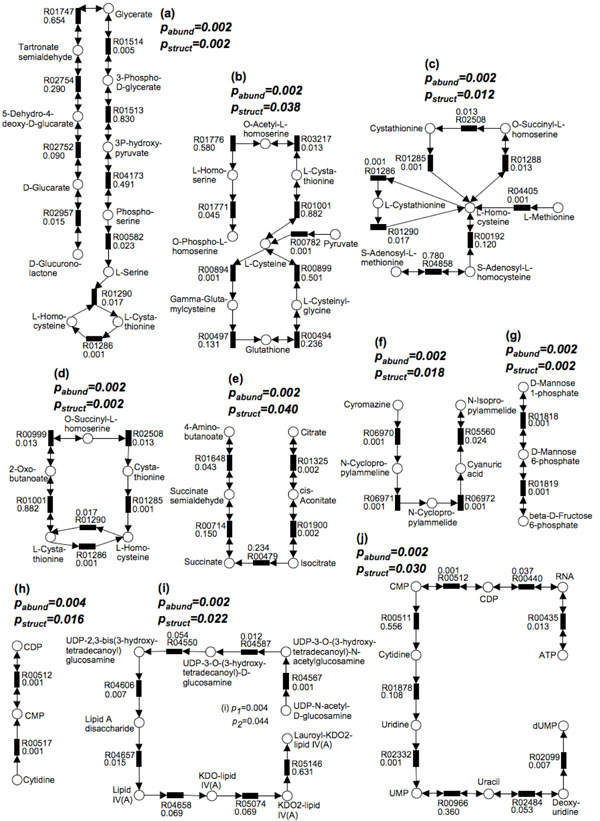
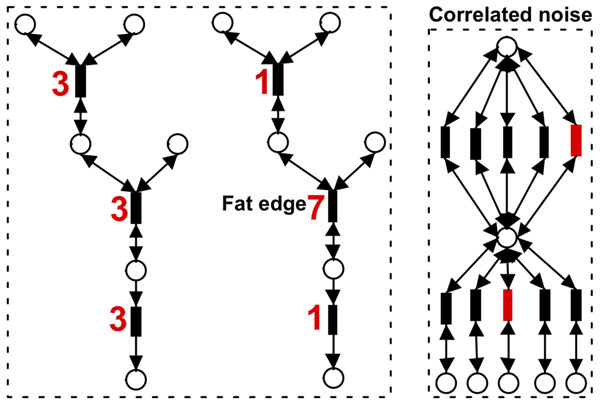
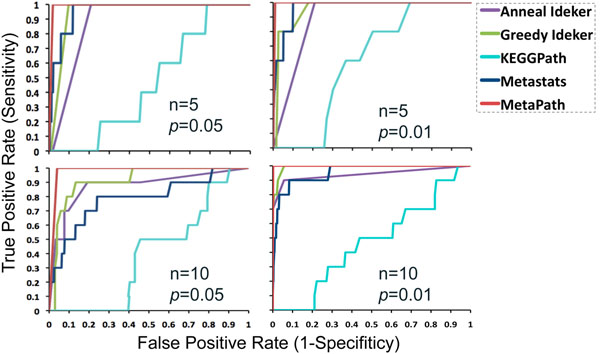
In order to validate our methods, we have designed a simulated metabolic pathways dataset and show that MetaPath outperforms other commonly used approaches. We also demonstrate the power of our methods in analyzing two publicly available metagenomic datasets, and show that the subnetworks identified by MetaPath provide valuable insights into the biological activities of the microbiome.
We have introduced a statistical method for finding significant metabolic subnetworks from metagenomic datasets. Compared with previous methods, results from MetaPath are more robust against noise in the data, and have significantly higher sensitivity and specificity (when tested on simulated datasets). When applied to two publicly available metagenomic datasets, the output of MetaPath is consistent with previous observations and also provides several new insights into the metabolic activity of the gut microbiome. The software is freely available at http://metapath.cbcb.umd.edu.
得益于测序技术的飞速发展,宏基因组学研究旨在对整个微生物群落进行特征描述,而无需培养单个细菌成员。宏基因组学研究的一个主要目标是确定微生物群落对其栖息地的特定功能适应性。通过将宏基因组序列映射到由数千个分子反应组成的全球代谢网络,可以估计样本的功能概况和丰度。在此,我们描述了一种强大的分析方法(MetaPath),该方法可以结合宏基因组序列数据和先前的代谢途径知识,识别宏基因组数据集中差异丰富的途径。
首先,我们为任意子网络引入一个评分函数,并通过贪婪搜索算法在全局网络中找到最大权重子网络。然后,我们使用非参数方法计算两个p值(pabund和pstruct),以回答两个不同的统计问题:(1)这个子网络的丰度是否有差异?(2)鉴于数据和网络结构,偶然发现如此好的子网络的概率是多少?最后,基于这两个p值发现显著的代谢子网络。
为了验证我们的方法,我们设计了一个模拟代谢途径数据集,并表明MetaPath优于其他常用方法。我们还展示了我们的方法在分析两个公开可用的宏基因组数据集方面的能力,并表明MetaPath识别出的子网络为微生物组的生物活性提供了有价值的见解。
我们引入了一种从宏基因组数据集中寻找显著代谢子网络的统计方法。与以前的方法相比,MetaPath的结果对数据中的噪声更具鲁棒性,并且在模拟数据集上测试时具有显著更高的灵敏度和特异性。当应用于两个公开可用的宏基因组数据集时,MetaPath的输出与先前的观察结果一致,并且还为肠道微生物组的代谢活性提供了一些新的见解。该软件可在http://metapath.cbcb.umd.edu免费获取。