Danko Charles G, Pertsov Arkady M
Department of Pharmacology, SUNY Upstate Medical University, Syracuse, NY, USA.
BMC Med Genomics. 2009 May 28;2:31. doi: 10.1186/1755-8794-2-31.
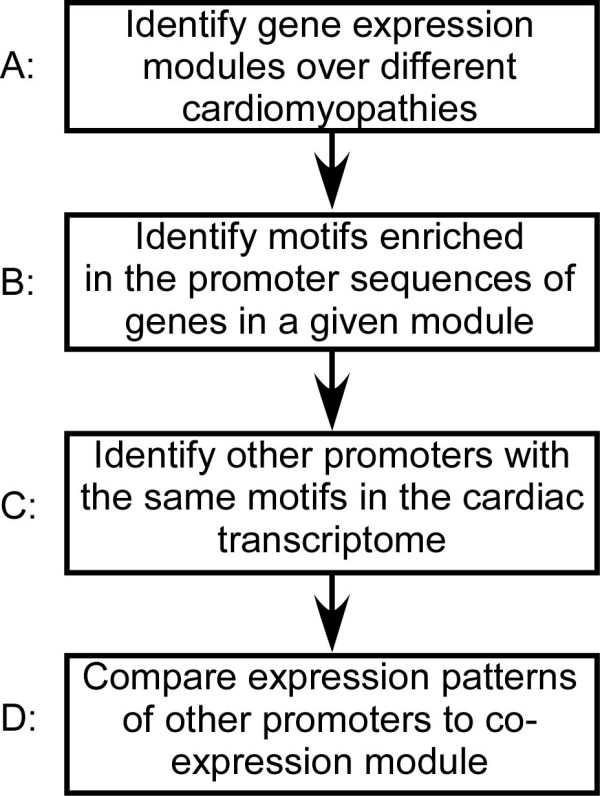
Cardiomyopathies, degenerative diseases of cardiac muscle, are among the leading causes of death in the developed world. Microarray studies of cardiomyopathies have identified up to several hundred genes that significantly alter their expression patterns as the disease progresses. However, the regulatory mechanisms driving these changes, in particular the networks of transcription factors involved, remain poorly understood. Our goals are (A) to identify modules of co-regulated genes that undergo similar changes in expression in various types of cardiomyopathies, and (B) to reveal the specific pattern of transcription factor binding sites, cis-elements, in the proximal promoter region of genes comprising such modules.
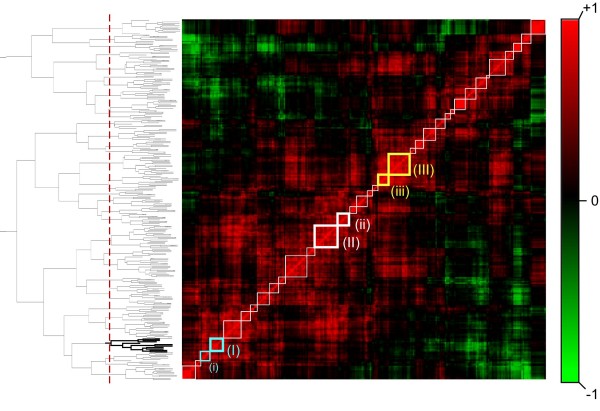
We analyzed 149 microarray samples from human hypertrophic and dilated cardiomyopathies of various etiologies. Hierarchical clustering and Gene Ontology annotations were applied to identify modules enriched in genes with highly correlated expression and a similar physiological function. To discover motifs that may underly changes in expression, we used the promoter regions for genes in three of the most interesting modules as input to motif discovery algorithms. The resulting motifs were used to construct a probabilistic model predictive of changes in expression across different cardiomyopathies.
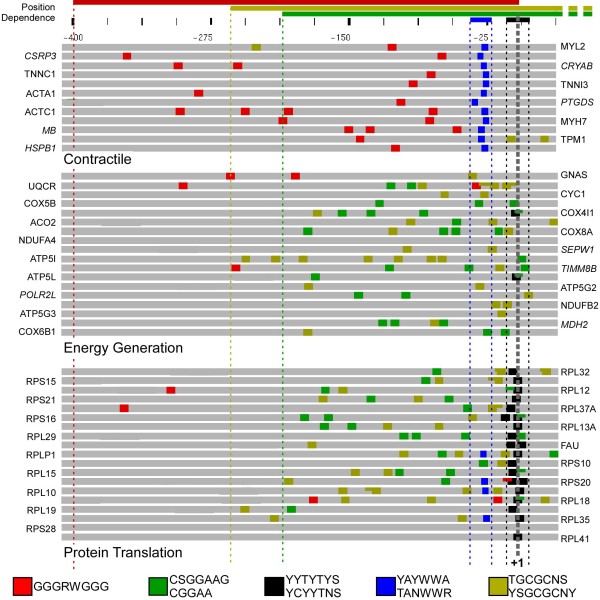
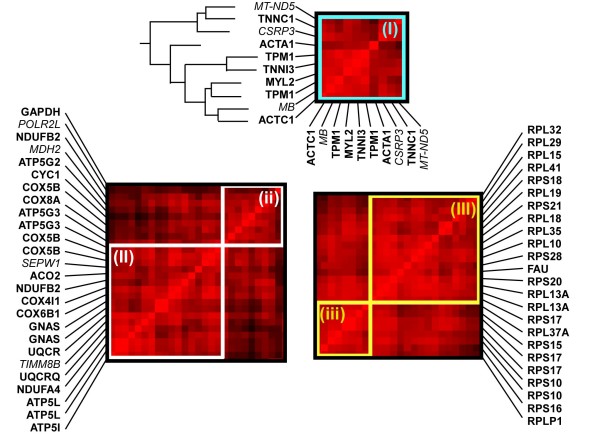
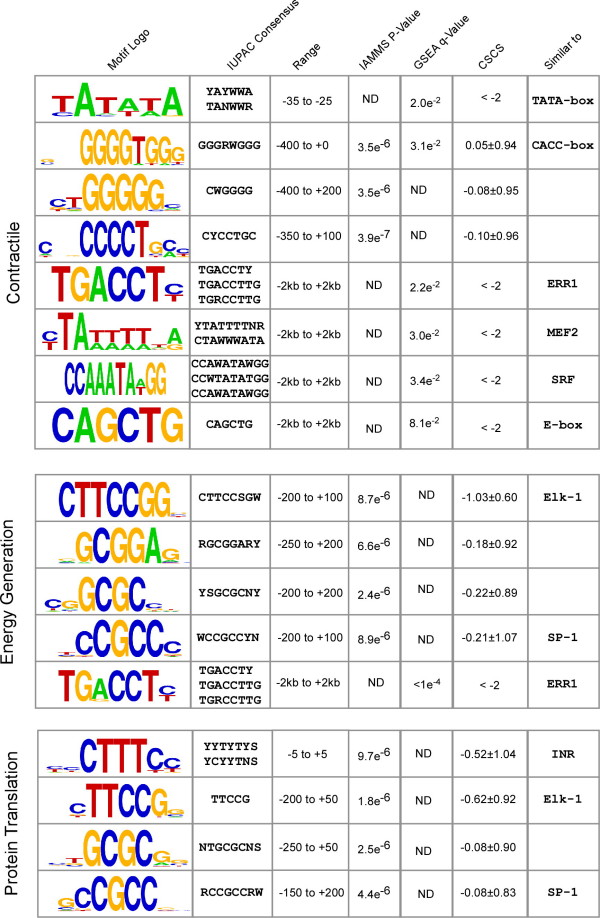
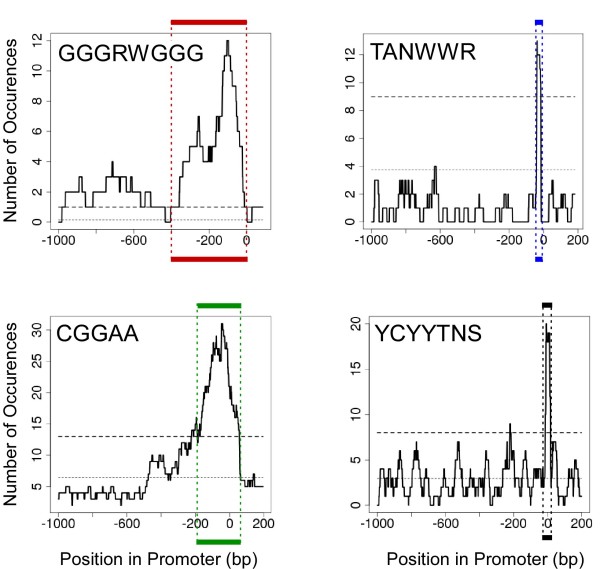
We found that three modules with the highest degree of functional enrichment contain genes involved in myocardial contraction (n = 9), energy generation (n = 20), or protein translation (n = 20). Using motif discovery tools revealed that genes in the contractile module were found to contain a TATA-box followed by a CACC-box, and are depleted in other GC-rich motifs; whereas genes in the translation module contain a pyrimidine-rich initiator, Elk-1, SP-1, and a novel motif with a GCGC core. Using a naïve Bayes classifier revealed that patterns of motifs are statistically predictive of expression patterns, with odds ratios of 2.7 (contractile), 1.9 (energy generation), and 5.5 (protein translation).
We identified patterns comprised of putative cis-regulatory motifs enriched in the upstream promoter sequence of genes that undergo similar changes in expression secondary to cardiomyopathies of various etiologies. Our analysis is a first step towards understanding transcription factor networks that are active in regulating gene expression during degenerative heart disease.
心肌病是心肌的退行性疾病,是发达国家主要的死亡原因之一。对心肌病的微阵列研究已确定多达数百个基因,随着疾病进展,这些基因的表达模式会发生显著改变。然而,驱动这些变化的调控机制,尤其是所涉及的转录因子网络,仍知之甚少。我们的目标是:(A)识别在各种类型心肌病中表达发生类似变化的共调控基因模块;(B)揭示构成此类模块的基因近端启动子区域中转录因子结合位点(顺式元件)的特定模式。
我们分析了来自各种病因的人类肥厚型和扩张型心肌病的149个微阵列样本。应用层次聚类和基因本体注释来识别富含表达高度相关且具有相似生理功能的基因的模块。为了发现可能是表达变化基础的基序,我们将三个最有趣模块中的基因启动子区域用作基序发现算法的输入。所得基序用于构建预测不同心肌病中表达变化的概率模型。
我们发现功能富集程度最高的三个模块包含参与心肌收缩(n = 9)、能量生成(n = 20)或蛋白质翻译(n = 20)的基因。使用基序发现工具发现,收缩模块中的基因含有一个TATA盒,后面跟着一个CACC盒,并且在其他富含GC的基序中含量较低;而翻译模块中的基因含有一个富含嘧啶的起始子、Elk-1、SP-1以及一个具有GCGC核心的新基序。使用朴素贝叶斯分类器发现,基序模式在统计学上可预测表达模式,收缩模块的优势比为2.7,能量生成模块为1.9,蛋白质翻译模块为5.5。
我们识别出了由假定的顺式调控基序组成的模式,这些基序富集在因各种病因导致的心肌病而表达发生类似变化的基因上游启动子序列中。我们的分析是迈向理解在退行性心脏病中调节基因表达的活跃转录因子网络的第一步。