Tsou Chih-Chiang, Tsui Yin-Hao, Yian Yi-Hwa, Chen Yi-Ju, Yang Han-Yin, Yu Chuan-Yih, Lynn Ke-Shiuan, Chen Yu-Ju, Sung Ting-Yi, Hsu Wen-Lian
Institute of Information Science and Institute of Chemistry, Academia Sinica, Nankang, Taipei, Taiwan 115.
Nucleic Acids Res. 2009 Jul;37(Web Server issue):W661-9. doi: 10.1093/nar/gkp476. Epub 2009 Jun 15.
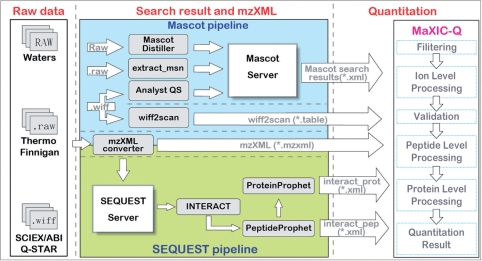
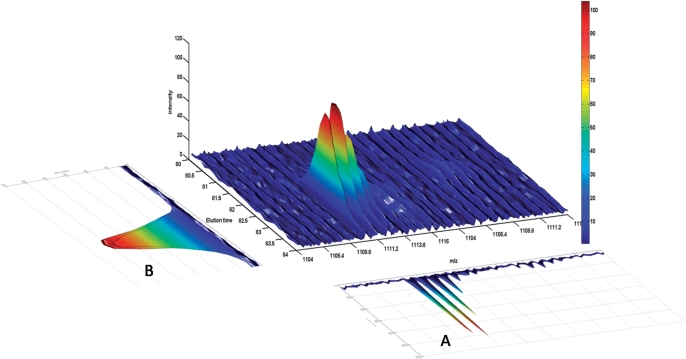
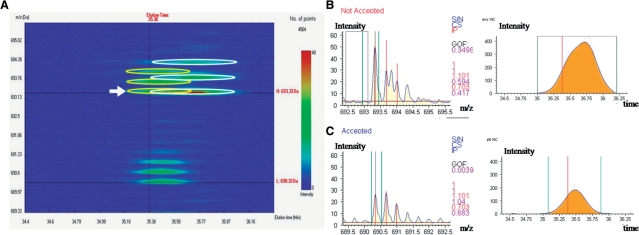
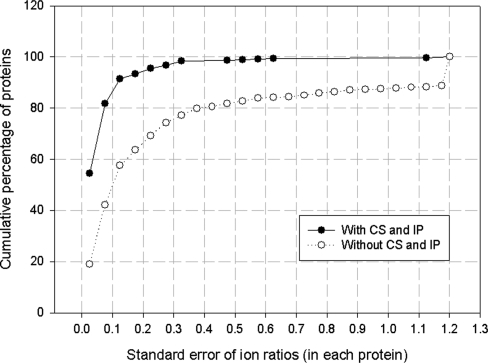
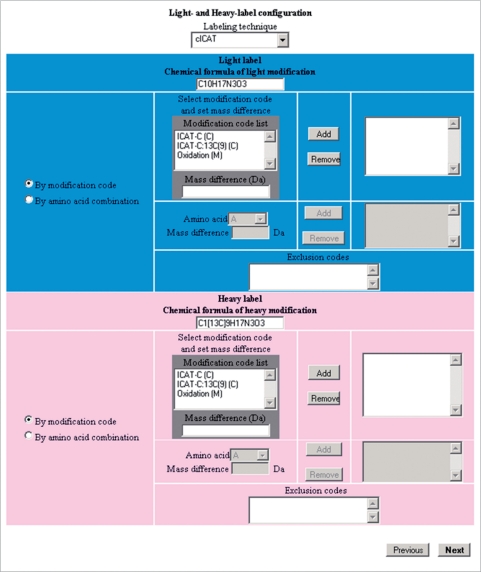
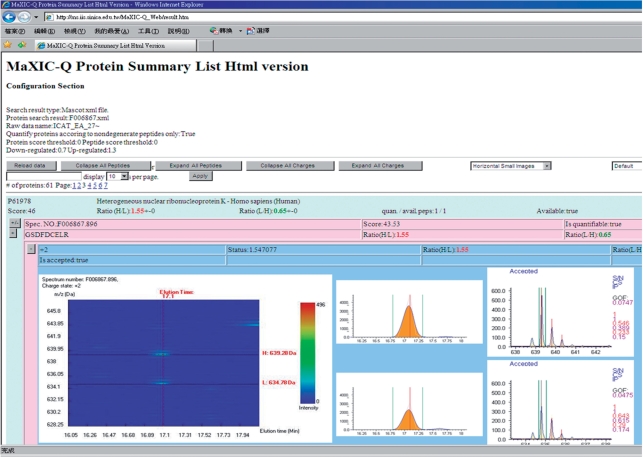
Isotope labeling combined with liquid chromatography-mass spectrometry (LC-MS) provides a robust platform for analyzing differential protein expression in proteomics research. We present a web service, called MaXIC-Q Web (http://ms.iis.sinica.edu.tw/MaXIC-Q_Web/), for quantitation analysis of large-scale datasets generated from proteomics experiments using various stable isotope-labeling techniques, e.g. SILAC, ICAT and user-developed labeling methods. It accepts spectral files in the standard mzXML format and search results from SEQUEST, Mascot and ProteinProphet as input. Furthermore, MaXIC-Q Web uses statistical and computational methods to construct two kinds of elution profiles for each ion, namely, PIMS (projected ion mass spectrum) and XIC (extracted ion chromatogram) from MS data. Toward accurate quantitation, a stringent validation procedure is performed on PIMSs to filter out peptide ions interfered with co-eluting peptides or noise. The areas of XICs determine ion abundances, which are used to calculate peptide and protein ratios. Since MaXIC-Q Web adopts stringent validation on spectral data, it achieves high accuracy so that manual validation effort can be substantially reduced. Furthermore, it provides various visualization diagrams and comprehensive quantitation reports so that users can conveniently inspect quantitation results. In summary, MaXIC-Q Web is a user-friendly, interactive, robust, generic web service for quantitation based on ICAT and SILAC labeling techniques.
同位素标记结合液相色谱-质谱联用(LC-MS)为蛋白质组学研究中的差异蛋白质表达分析提供了一个强大的平台。我们展示了一个名为MaXIC-Q Web(http://ms.iis.sinica.edu.tw/MaXIC-Q_Web/)的网络服务,用于对使用各种稳定同位素标记技术(如SILAC、ICAT和用户开发的标记方法)从蛋白质组学实验生成的大规模数据集进行定量分析。它接受标准mzXML格式的光谱文件以及来自SEQUEST、Mascot和ProteinProphet的搜索结果作为输入。此外,MaXIC-Q Web使用统计和计算方法为每个离子构建两种洗脱图谱,即来自质谱数据的投影离子质谱(PIMS)和提取离子色谱图(XIC)。为了实现准确的定量,对PIMS执行严格的验证程序,以滤除受共洗脱肽或噪声干扰的肽离子。XIC的面积决定离子丰度,用于计算肽和蛋白质的比率。由于MaXIC-Q Web对光谱数据采用严格的验证,它具有很高的准确性,从而可以大幅减少人工验证工作。此外,它提供各种可视化图表和全面的定量报告,以便用户可以方便地检查定量结果。总之,MaXIC-Q Web是一个基于ICAT和SILAC标记技术的用户友好、交互式、强大且通用的定量网络服务。