Tritchler David, Parkhomenko Elena, Beyene Joseph
Department of Biostatistics, University of Toronto, Toronto, Ontario, Canada.
BMC Bioinformatics. 2009 Jun 23;10:193. doi: 10.1186/1471-2105-10-193.
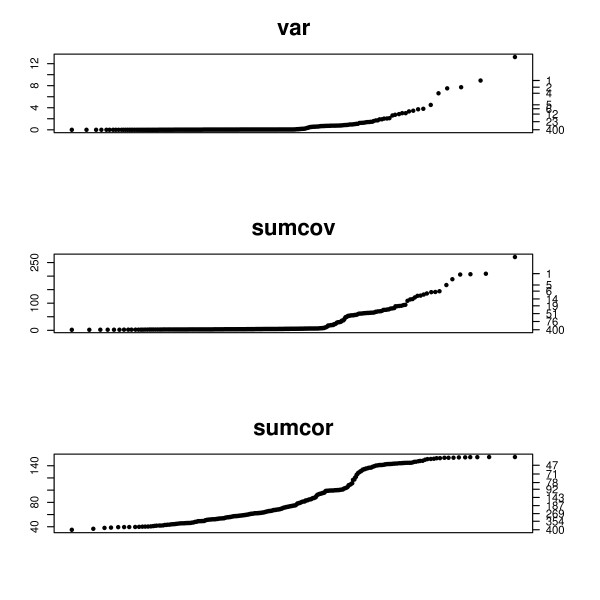
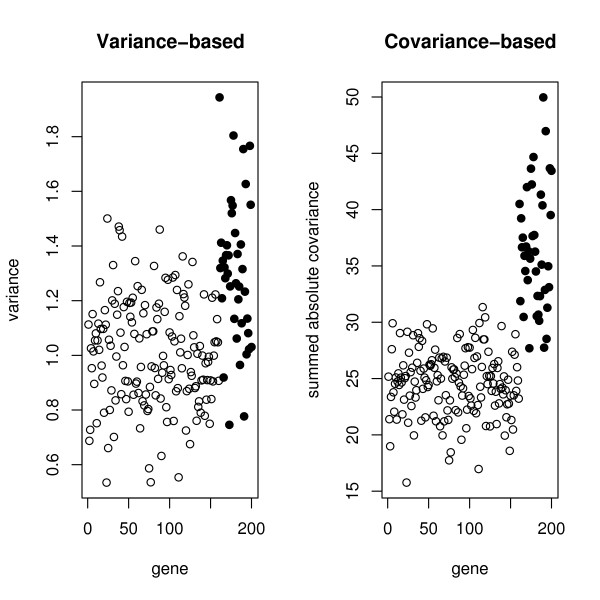
Prior to cluster analysis or genetic network analysis it is customary to filter, or remove genes considered to be irrelevant from the set of genes to be analyzed. Often genes whose variation across samples is less than an arbitrary threshold value are deleted. This can improve interpretability and reduce bias.
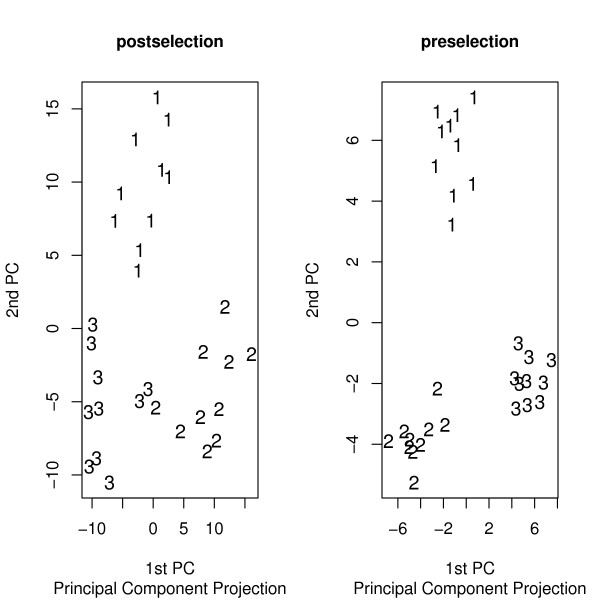
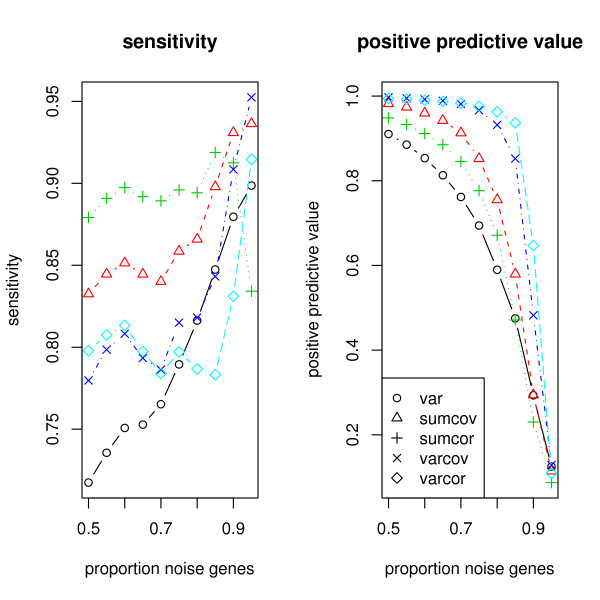
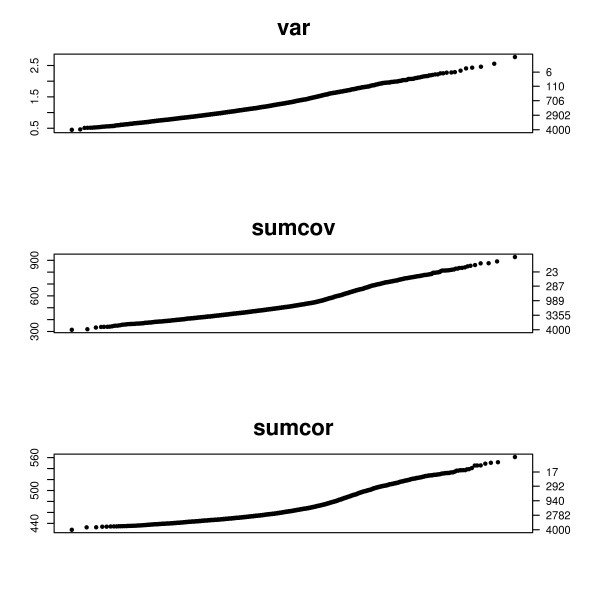
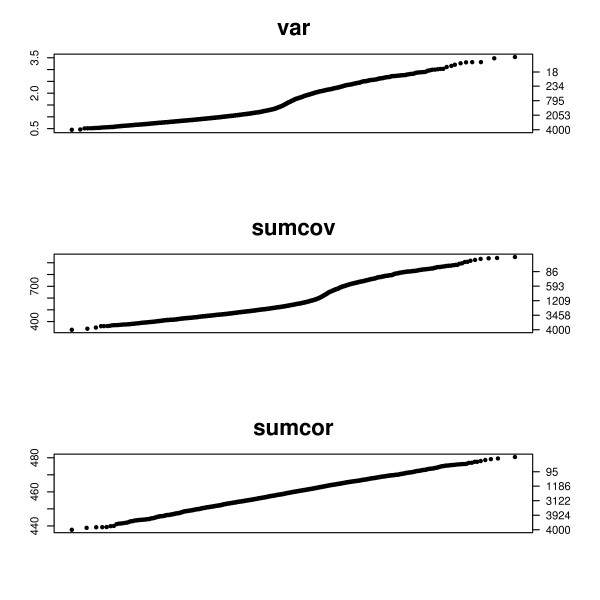
This paper introduces modular models for representing network structure in order to study the relative effects of different filtering methods. We show that cluster analysis and principal components are strongly affected by filtering. Filtering methods intended specifically for cluster and network analysis are introduced and compared by simulating modular networks with known statistical properties. To study more realistic situations, we analyze simulated "real" data based on well-characterized E. coli and S. cerevisiae regulatory networks.
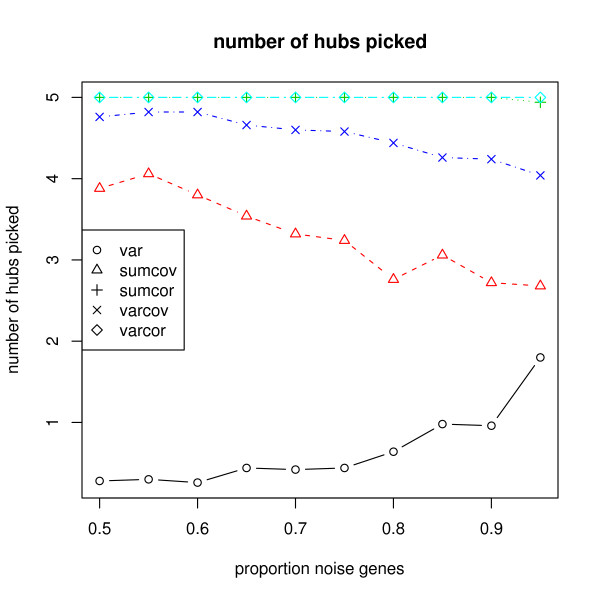
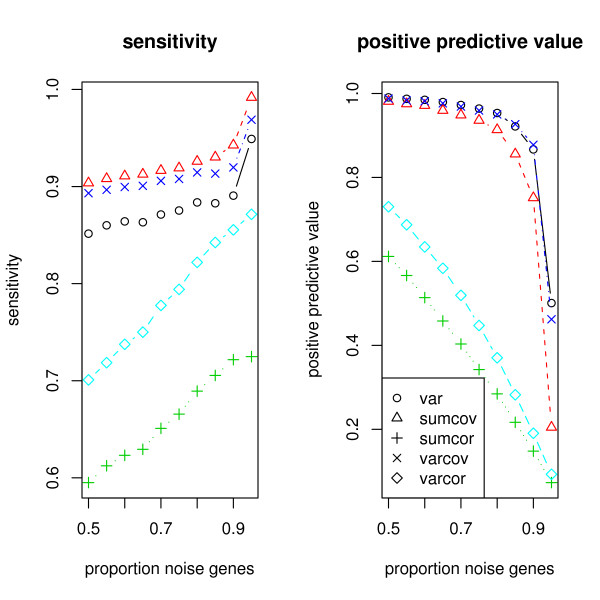
The methods introduced apply very generally, to any similarity matrix describing gene expression. One of the proposed methods, SUMCOV, performed well for all models simulated.
在进行聚类分析或基因网络分析之前,通常会从待分析的基因集中筛选或去除被认为无关的基因。通常会删除那些在样本间变异小于任意阈值的基因。这可以提高可解释性并减少偏差。
本文引入了用于表示网络结构的模块化模型,以研究不同筛选方法的相对影响。我们表明聚类分析和主成分分析受筛选的影响很大。通过模拟具有已知统计特性的模块化网络,引入并比较了专门用于聚类和网络分析的筛选方法。为了研究更现实的情况,我们基于特征明确的大肠杆菌和酿酒酵母调控网络分析了模拟的“真实”数据。
所介绍的方法非常通用,适用于描述基因表达的任何相似性矩阵。所提出的方法之一SUMCOV在所有模拟模型中表现良好。