Wiesner Magdalena, Zaidi Mussaret B, Calva Edmundo, Fernández-Mora Marcos, Calva Juan J, Silva Claudia
Departamento de Microbiología Molecular, Instituto de Biotecnología, Universidad Nacional Autónoma de México, Cuernavaca, México.
BMC Microbiol. 2009 Jul 3;9:131. doi: 10.1186/1471-2180-9-131.
Bacterial genomes are mosaic structures composed of genes present in every strain of the same species (core genome), and genes present in some but not all strains of a species (accessory genome). The aim of this study was to compare the genetic diversity of core and accessory genes of a Salmonella enterica subspecies enterica serovar Typhimurium (Typhimurium) population isolated from food-animal and human sources in four regions of Mexico. Multilocus sequence typing (MLST) and macrorestriction fingerprints by pulsed-field gel electrophoresis (PFGE) were used to address the core genetic variation, and genes involved in pathogenesis and antibiotic resistance were selected to evaluate the accessory genome.
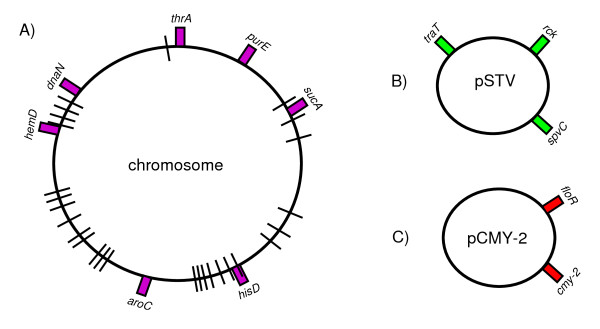
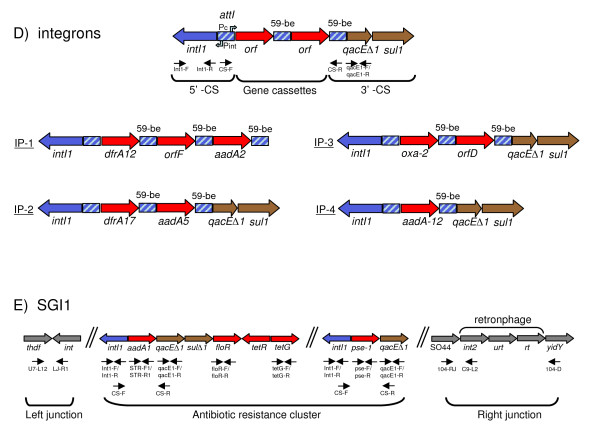
We found a low genetic diversity for both housekeeping and accessory genes. Sequence type 19 (ST19) was supported as the founder genotype of STs 213, 302 and 429. We found a temporal pattern in which the derived ST213 is replacing the founder ST19 in the four geographic regions analyzed and a geographic trend in the number of resistance determinants. The distribution of the accessory genes was not random among chromosomal genotypes. We detected strong associations among the different accessory genes and the multilocus chromosomal genotypes (STs). First, the Salmonella virulence plasmid (pSTV) was found mostly in ST19 isolates. Second, the plasmid-borne betalactamase cmy-2 was found only in ST213 isolates. Third, the most abundant integron, IP-1 (dfrA12, orfF and aadA2), was found only in ST213 isolates. Fourth, the Salmonella genomic island (SGI1) was found mainly in a subgroup of ST19 isolates carrying pSTV. The mapping of accessory genes and multilocus genotypes on the dendrogram derived from macrorestiction fingerprints allowed the establishment of genetic subgroups within the population.
Despite the low levels of genetic diversity of core and accessory genes, the non-random distribution of the accessory genes across chromosomal backgrounds allowed us to discover genetic subgroups within the population. This study provides information about the importance of the accessory genome in generating genetic variability within a bacterial population.
细菌基因组是镶嵌结构,由同一物种每个菌株中都存在的基因(核心基因组)以及一个物种中部分但并非所有菌株中存在的基因(辅助基因组)组成。本研究的目的是比较从墨西哥四个地区的食用动物和人类来源分离的肠炎沙门氏菌肠炎亚种鼠伤寒血清型(鼠伤寒沙门氏菌)群体的核心基因和辅助基因的遗传多样性。采用多位点序列分型(MLST)和脉冲场凝胶电泳(PFGE)的宏观限制性指纹图谱来研究核心遗传变异,并选择参与致病机制和抗生素抗性的基因来评估辅助基因组。
我们发现看家基因和辅助基因的遗传多样性都很低。序列型19(ST19)被确认为ST213、302和429的奠基者基因型。我们发现了一种时间模式,即在分析的四个地理区域中,衍生的ST213正在取代奠基者ST19,并且在抗性决定因素的数量上存在地理趋势。辅助基因在染色体基因型中的分布并非随机。我们检测到不同辅助基因与多位点染色体基因型(STs)之间存在强关联。首先,沙门氏菌毒力质粒(pSTV)大多在ST19分离株中发现。其次,质粒携带的β-内酰胺酶cmy-2仅在ST213分离株中发现。第三,最丰富的整合子IP-1(dfrA12、orfF和aadA2)仅在ST213分离株中发现。第四,沙门氏菌基因组岛(SGI1)主要在携带pSTV的ST19分离株亚组中发现。将辅助基因和多位点基因型映射到由宏观限制性指纹图谱得出的树状图上,使得能够在群体中建立遗传亚组。
尽管核心基因和辅助基因的遗传多样性水平较低,但辅助基因在染色体背景中的非随机分布使我们能够在群体中发现遗传亚组。本研究提供了关于辅助基因组在细菌群体中产生遗传变异性方面重要性的信息。