Department of Pharmacology and Toxicology, University of Utah, 112 Skaggs Hall, Salt Lake City, UT 84112, USA.
Breast Cancer Res. 2009;11(4):R55. doi: 10.1186/bcr2344. Epub 2009 Jul 28.
Perhaps the major challenge in developing more effective therapeutic strategies for the treatment of breast cancer patients is confronting the heterogeneity of the disease, recognizing that breast cancer is not one disease but multiple disorders with distinct underlying mechanisms. Gene-expression profiling studies have been used to dissect this complexity, and our previous studies identified a series of intrinsic subtypes of breast cancer that define distinct populations of patients with respect to survival. Additional work has also used signatures of oncogenic pathway deregulation to dissect breast cancer heterogeneity as well as to suggest therapeutic opportunities linked to pathway activation.
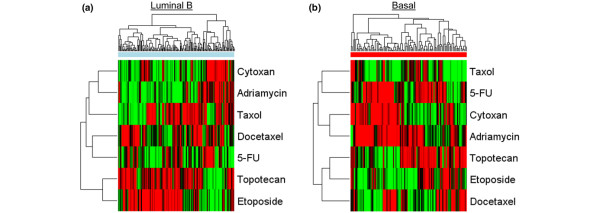
We used genomic analyses to identify relations between breast cancer subtypes, pathway deregulation, and drug sensitivity. For these studies, we use three independent breast cancer gene-expression data sets to measure an individual tumor phenotype. Correlation between pathway status and subtype are examined and linked to predictions for response to conventional chemotherapies.
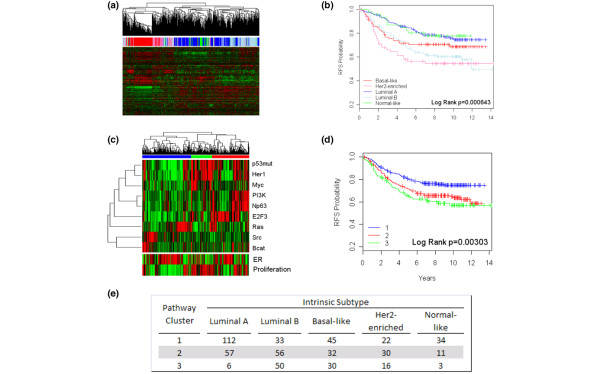
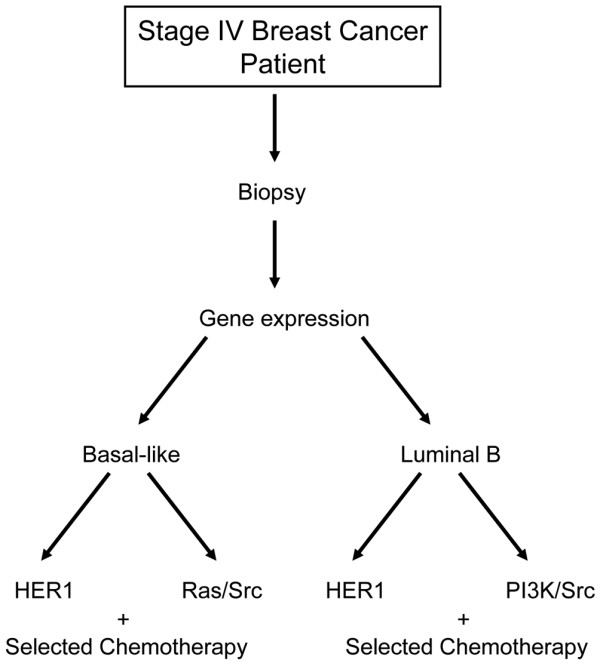
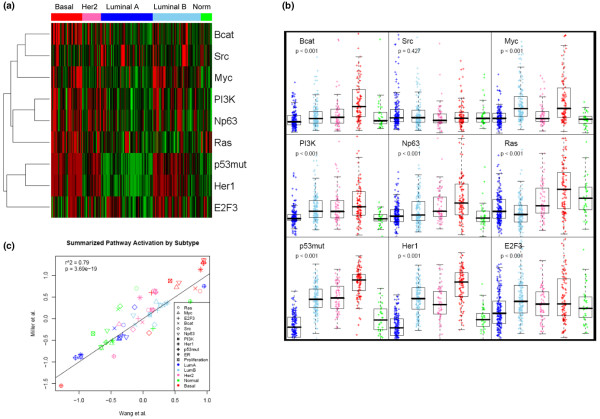
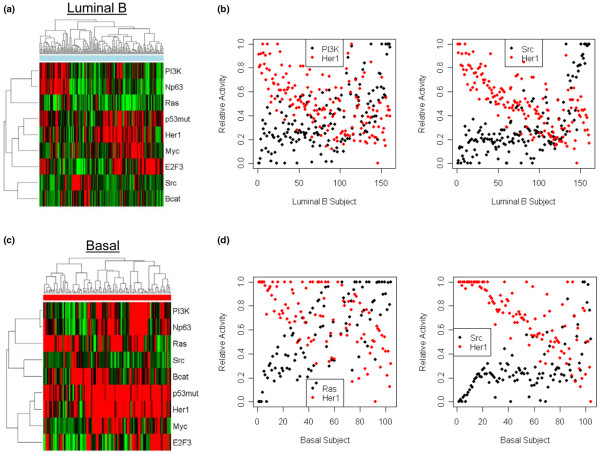
We reveal patterns of pathway activation characteristic of each molecular breast cancer subtype, including within the more aggressive subtypes in which novel therapeutic opportunities are critically needed. Whereas some oncogenic pathways have high correlations to breast cancer subtype (RAS, CTNNB1, p53, HER1), others have high variability of activity within a specific subtype (MYC, E2F3, SRC), reflecting biology independent of common clinical factors. Additionally, we combined these analyses with predictions of sensitivity to commonly used cytotoxic chemotherapies to provide additional opportunities for therapeutics specific to the intrinsic subtype that might be better aligned with the characteristics of the individual patient.
Genomic analyses can be used to dissect the heterogeneity of breast cancer. We use an integrated analysis of breast cancer that combines independent methods of genomic analyses to highlight the complexity of signaling pathways underlying different breast cancer phenotypes and to identify optimal therapeutic opportunities.
也许开发更有效的治疗策略来治疗乳腺癌患者的主要挑战是面对疾病的异质性,认识到乳腺癌不是一种疾病,而是具有不同潜在机制的多种疾病。基因表达谱研究已被用于剖析这种复杂性,我们之前的研究确定了一系列乳腺癌的内在亚型,这些亚型定义了在生存方面具有明显不同的患者群体。此外,其他研究还使用致癌途径失调的特征来剖析乳腺癌的异质性,并提出与途径激活相关的治疗机会。
我们使用基因组分析来确定乳腺癌亚型、途径失调和药物敏感性之间的关系。对于这些研究,我们使用三个独立的乳腺癌基因表达数据集来测量个体肿瘤表型。我们检查了途径状态与亚型之间的相关性,并将其与对常规化疗的反应预测联系起来。
我们揭示了每个分子乳腺癌亚型的特征性途径激活模式,包括在更具侵袭性的亚型中,这些亚型迫切需要新的治疗机会。虽然某些致癌途径与乳腺癌亚型高度相关(RAS、CTNNB1、p53、HER1),但其他途径在特定亚型内具有高度的活性变异性(MYC、E2F3、SRC),反映了独立于常见临床因素的生物学。此外,我们将这些分析与对常用细胞毒性化疗药物敏感性的预测相结合,为针对内在亚型的治疗提供了额外的机会,这些治疗可能与个体患者的特征更匹配。
基因组分析可用于剖析乳腺癌的异质性。我们使用乳腺癌的综合分析,结合基因组分析的独立方法,突出了不同乳腺癌表型背后信号通路的复杂性,并确定了最佳的治疗机会。