Bruni Roberto, Costantino Angela, Tritarelli Elena, Marcantonio Cinzia, Ciccozzi Massimo, Rapicetta Maria, El Sawaf Gamal, Giuliani Alessandro, Ciccaglione Anna Rita
Department of Infectious, Parasitic and Immune-mediated Diseases, Istituto Superiore di Sanità, Rome, Italy.
BMC Struct Biol. 2009 Jul 29;9:48. doi: 10.1186/1472-6807-9-48.
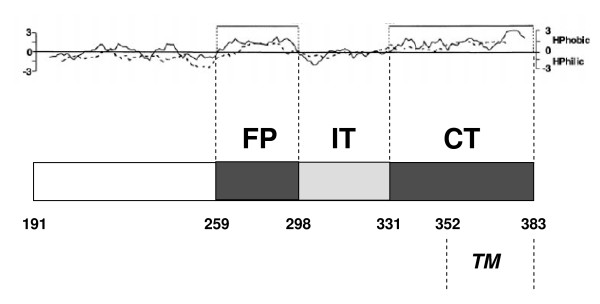
The E1 protein of Hepatitis C Virus (HCV) can be dissected into two distinct hydrophobic regions: a central domain containing an hypothetical fusion peptide (FP), and a C-terminal domain (CT) comprising two segments, a pre-anchor and a trans-membrane (TM) region. In the currently accepted model of the viral fusion process, the FP and the TM regions are considered to be closely juxtaposed in the post-fusion structure and their physical interaction cannot be excluded. In the present study, we took advantage of the natural sequence variability present among HCV strains to test, by purely sequence-based computational tools, the hypothesis that in this virus the fusion process involves the physical interaction of the FP and CT regions of E1.
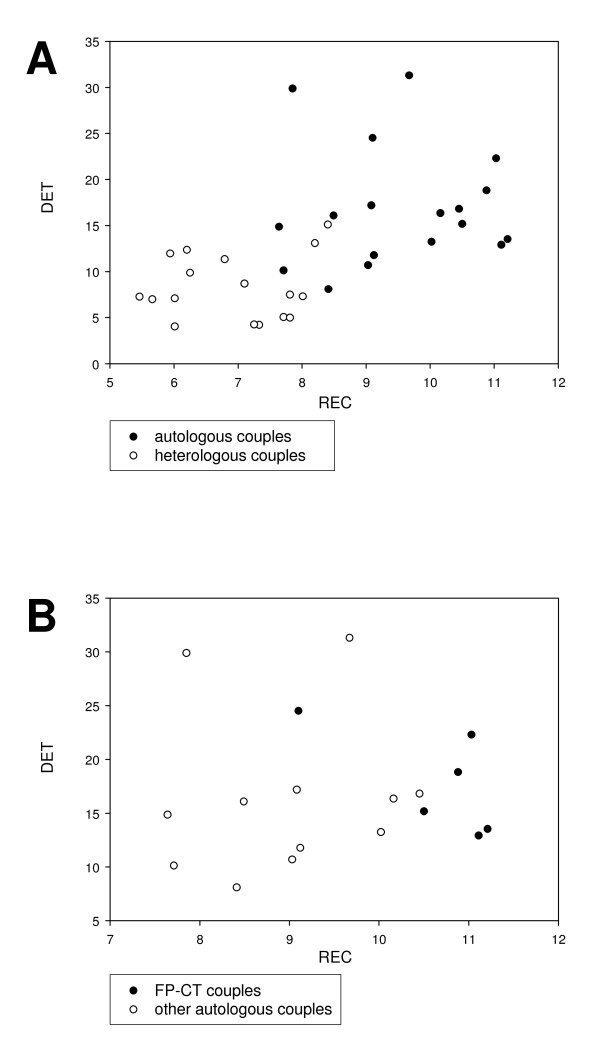
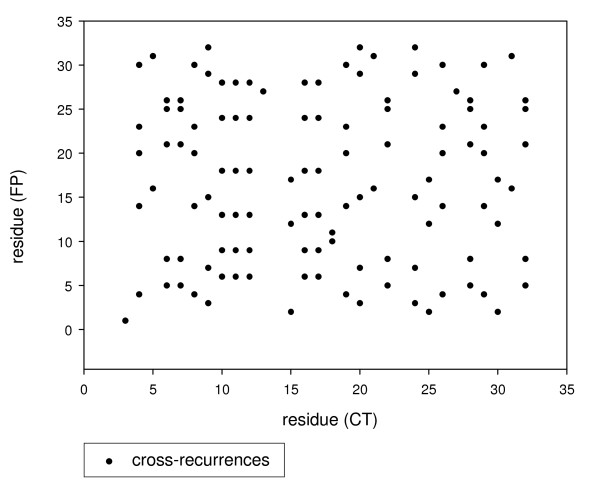
Two computational approaches were applied. The first one is based on the co-evolution paradigm of interacting peptides and consequently on the correlation between the distance matrices generated by the sequence alignment method applied to FP and CT primary structures, respectively. In spite of the relatively low random genetic drift between genotypes, co-evolution analysis of sequences from five HCV genotypes revealed a greater correlation between the FP and CT domains than respect to a control HCV sequence from Core protein, so giving a clear, albeit still inconclusive, support to the physical interaction hypothesis.The second approach relies upon a non-linear signal analysis method widely used in protein science called Recurrence Quantification Analysis (RQA). This method allows for a direct comparison of domains for the presence of common hydrophobicity patterns, on which the physical interaction is based upon. RQA greatly strengthened the reliability of the hypothesis by the scoring of a lot of cross-recurrences between FP and CT peptides hydrophobicity patterning largely outnumbering chance expectations and pointing to putative interaction sites. Intriguingly, mutations in the CT region of E1, reducing the fusion process in vitro, strongly reduced the amount of cross-recurrence further supporting interaction between this region and FP.
Our results support a fusion model for HCV in which the FP and the C-terminal region of E1 are juxtaposed and interact in the post-fusion structure. These findings have general implications for viruses, as any visualization of the post-fusion FP-TM complex has been precluded by the impossibility to obtain crystallised viral fusion proteins containing the trans-membrane region. This limitation gives to sequence based modelling efforts a crucial role in the sketching of a molecular interpretation of the fusion process. Moreover, our data also have a more general relevance for cell biology as the mechanism of intracellular fusion showed remarkable similarities with viral fusion.
丙型肝炎病毒(HCV)的E1蛋白可分为两个不同的疏水区域:一个包含假定融合肽(FP)的中央结构域,以及一个由两段组成的C端结构域(CT),即前锚定区和跨膜(TM)区。在目前被广泛接受的病毒融合过程模型中,FP和TM区在融合后结构中被认为紧密相邻,它们之间的物理相互作用也不能被排除。在本研究中,我们利用HCV毒株间存在的天然序列变异性,通过纯基于序列的计算工具,来检验E1的融合过程涉及FP和CT区物理相互作用这一假说。
我们应用了两种计算方法。第一种基于相互作用肽的协同进化范式,因此基于分别应用于FP和CT一级结构的序列比对方法所生成的距离矩阵之间的相关性。尽管不同基因型之间的随机遗传漂变相对较低,但对五种HCV基因型序列的协同进化分析显示,FP和CT结构域之间的相关性高于核心蛋白的对照HCV序列,从而为物理相互作用假说提供了明确但仍不确定的支持。第二种方法依赖于蛋白质科学中广泛使用的一种非线性信号分析方法,称为递归量化分析(RQA)。该方法允许直接比较各结构域是否存在基于物理相互作用的共同疏水性模式。RQA通过对FP和CT肽疏水性模式之间大量交叉递归的评分,大大增强了该假说的可靠性,这些交叉递归的数量大大超过了随机预期,并指向了假定的相互作用位点。有趣的是,E1的CT区发生突变,在体外降低了融合过程,同时也大大减少了交叉递归的数量,进一步支持了该区域与FP之间的相互作用。
我们的结果支持HCV的一种融合模型,其中E1的FP和C端区域在融合后结构中并列且相互作用。这些发现对病毒具有普遍意义,因为由于无法获得包含跨膜区域的结晶病毒融合蛋白,融合后FP-TM复合物的任何可视化研究都受到了阻碍。这一局限性使得基于序列的建模工作在勾勒融合过程的分子解释中发挥关键作用。此外,我们的数据对细胞生物学也具有更广泛的相关性,因为细胞内融合机制与病毒融合表现出显著的相似性。