Département Santé des Plantes et Environnement, Institut National de la Recherche Agronomique, Beaucouzé, France.
PLoS One. 2009 Aug 14;4(8):e6632. doi: 10.1371/journal.pone.0006632.
The genetic basis of host specificity for animal and plant pathogenic bacteria remains poorly understood. For plant pathogenic bacteria, host range is restricted to one or a few host plant species reflecting a tight adaptation to specific hosts.
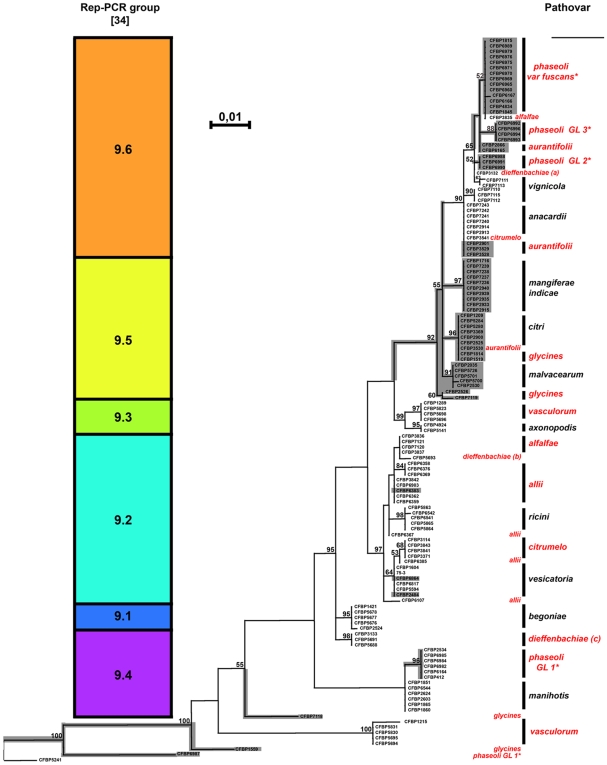
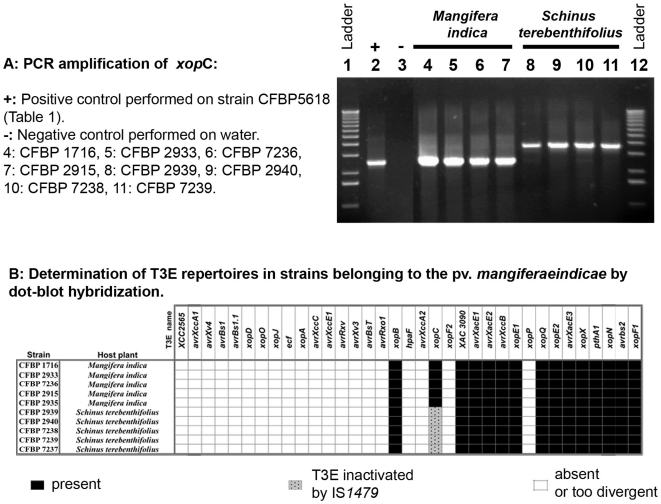
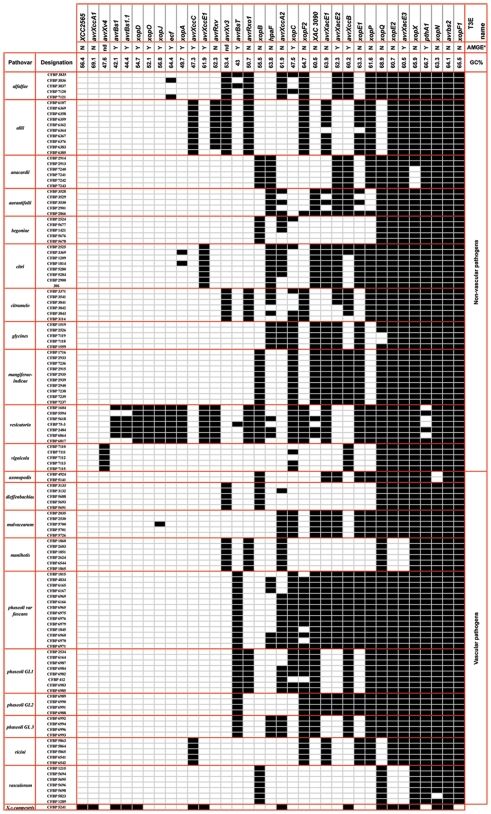
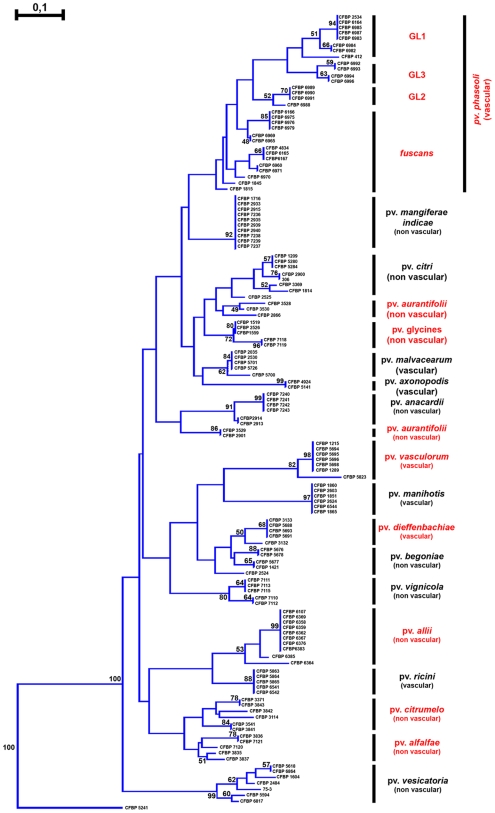
METHODOLOGY/PRINCIPAL FINDINGS: Two hypotheses can be formulated to explain host specificity: either it can be explained by the phylogenetic position of the strains, or by the association of virulence genes enabling a pathological convergence of phylogenically distant strains. In this latter hypothesis, host specificity would result from the interaction between repertoires of bacterial virulence genes and repertoires of genes involved in host defences. To challenge these two hypotheses, we selected 132 Xanthomonas axonopodis strains representative of 18 different pathovars which display different host range. First, the phylogenetic position of each strain was determined by sequencing the housekeeping gene rpoD. This study showed that many pathovars of Xanthomonas axonopodis are polyphyletic. Second, we investigated the distribution of 35 type III effector genes (T3Es) in these strains by both PCR and hybridization methods. Indeed, for pathogenic bacteria T3Es were shown to trigger and to subvert host defences. Our study revealed that T3E repertoires comprise core and variable gene suites that likely have distinct roles in pathogenicity and different evolutionary histories. Our results showed a correspondence between composition of T3E repertoires and pathovars of Xanthomonas axonopodis. For polyphyletic pathovars, this suggests that T3E genes might explain a pathological convergence of phylogenetically distant strains. We also identified several DNA rearrangements within T3E genes, some of which correlate with host specificity of strains.
CONCLUSIONS/SIGNIFICANCE: These data provide insight into the potential role played by T3E genes for pathogenic bacteria and support a "repertoire for repertoire" hypothesis that may explain host specificity. Our work provides resources for functional and evolutionary studies aiming at understanding host specificity of pathogenic bacteria, functional redundancy between T3Es and the driving forces shaping T3E repertoires.
动物和植物病原菌宿主特异性的遗传基础仍知之甚少。对于植物病原菌,其宿主范围局限于一种或几种宿主植物物种,反映了对特定宿主的紧密适应。
方法/主要发现:可以提出两种假说来解释宿主特异性:一种可以用菌株的系统发育位置来解释,另一种可以用毒力基因的关联来解释,这些基因使系统发育上遥远的菌株在病理上趋同。在后一种假说中,宿主特异性是由细菌毒力基因库与宿主防御基因库之间的相互作用产生的。为了挑战这两种假说,我们选择了 132 株代表 18 种不同致病变种的黄单胞菌 axonopodis 菌株,这些菌株表现出不同的宿主范围。首先,通过测序管家基因 rpoD 来确定每个菌株的系统发育位置。这项研究表明,许多黄单胞菌 axonopodis 的致病变种是多系的。其次,我们通过 PCR 和杂交方法研究了这些菌株中 35 种 III 型效应基因 (T3E) 的分布。事实上,对于病原菌来说,T3E 被证明可以触发和颠覆宿主防御。我们的研究表明,T3E 基因库由核心和可变基因套件组成,这些基因套件可能在致病性方面具有不同的作用和不同的进化历史。我们的研究结果表明,T3E 基因库的组成与黄单胞菌 axonopodis 的致病变种之间存在对应关系。对于多系致病变种,这表明 T3E 基因可能解释了系统发育上遥远的菌株在病理上的趋同。我们还在 T3E 基因内发现了几种 DNA 重排,其中一些与菌株的宿主特异性相关。
结论/意义:这些数据为 T3E 基因在病原菌中的潜在作用提供了深入了解,并支持了一个“基因库对基因库”的假说,该假说可能解释宿主特异性。我们的工作为功能和进化研究提供了资源,旨在理解病原菌的宿主特异性、T3E 之间的功能冗余以及塑造 T3E 基因库的驱动力。