Morris K Udall Parkinson's Disease Research Center of Excellence, University of Virginia School of Medicine, Charlottesville, VA 22908, USA.
Mol Neurodegener. 2009 Sep 23;4:37. doi: 10.1186/1750-1326-4-37.
Sporadic Parkinson's disease (sPD) is a nervous system-wide disease that presents with a bradykinetic movement disorder and is frequently complicated by depression and cognitive impairment. sPD likely has multiple interacting causes that include increased oxidative stress damage to mitochondrial components and reduced mitochondrial bioenergetic capacity. We analyzed mitochondria from postmortem sPD and CTL brains for evidence of oxidative damage to mitochondrial DNA (mtDNA), heteroplasmic mtDNA point mutations and levels of electron transport chain proteins. We sought to determine if sPD brains possess any mtDNA genotype-respiratory phenotype relationships.
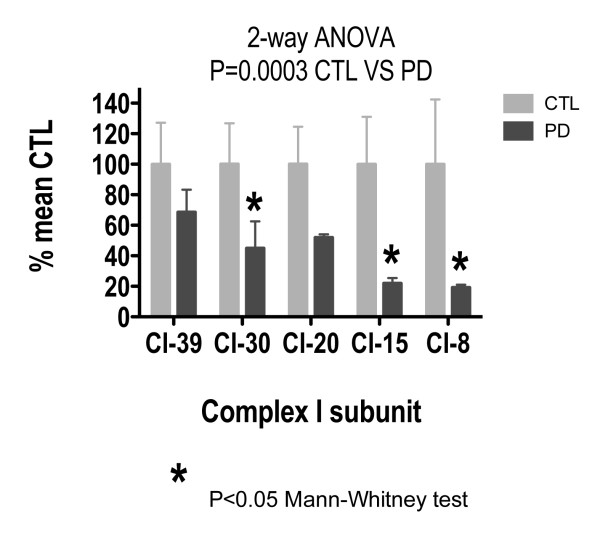
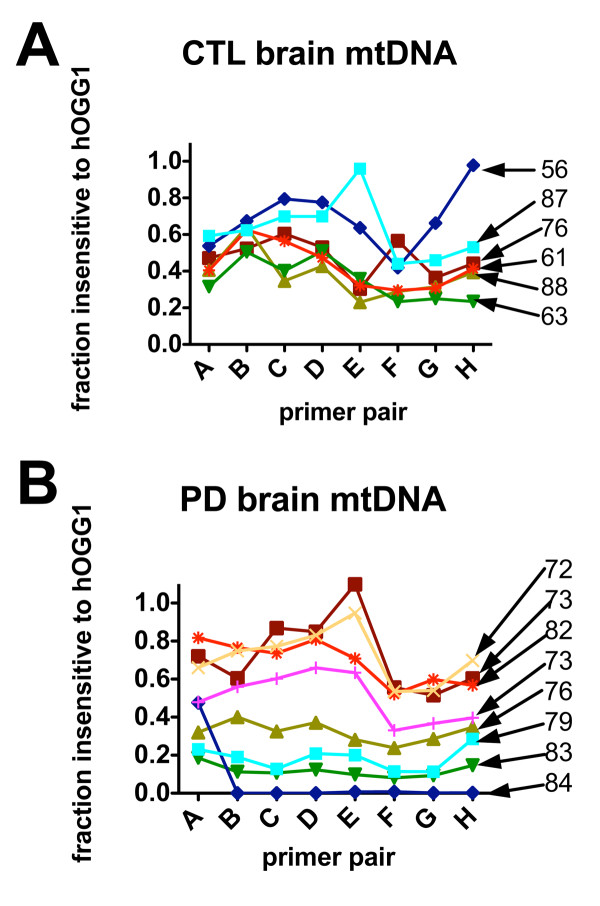
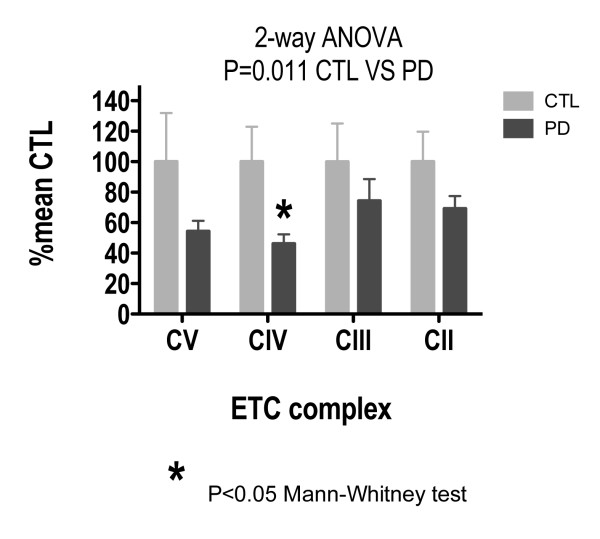
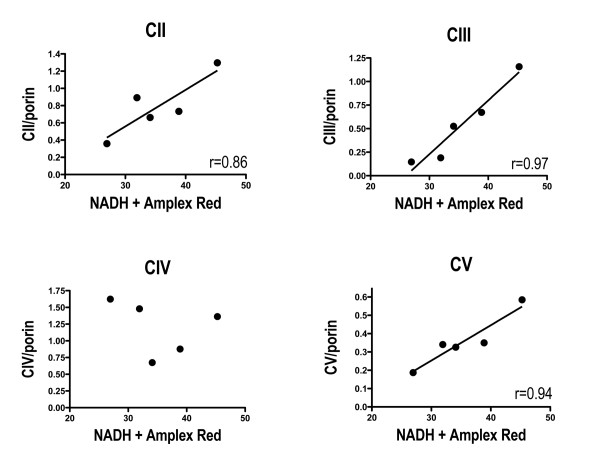
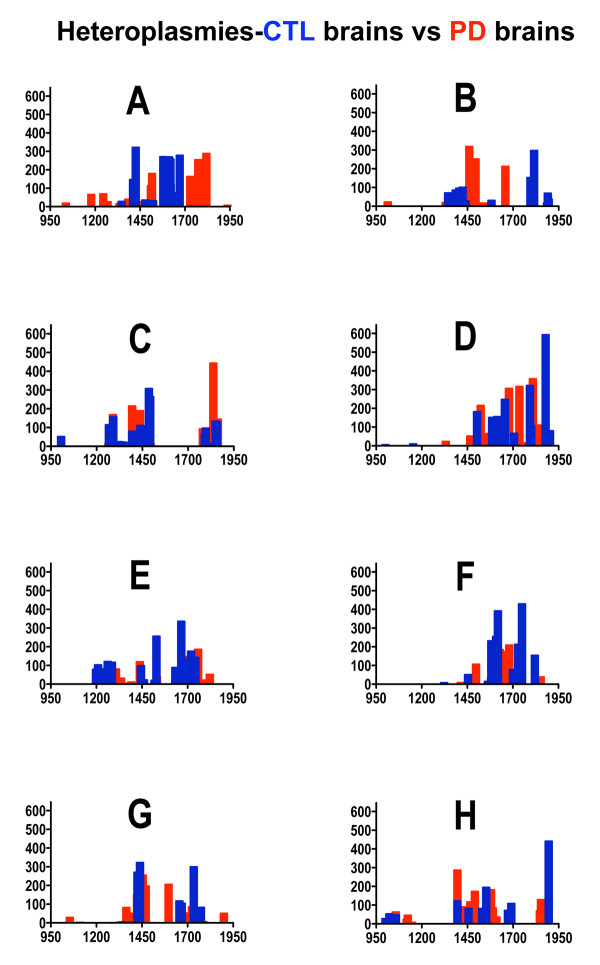
Treatment of sPD brain mtDNA with the mitochondrial base-excision repair enzyme 8-oxyguanosine glycosylase-1 (hOGG1) inhibited, in an age-dependent manner, qPCR amplification of overlapping ~2 kbase products; amplification of CTL brain mtDNA showed moderate sensitivity to hOGG1 not dependent on donor age. hOGG1 mRNA expression was not different between sPD and CTL brains. Heteroplasmy analysis of brain mtDNA using Surveyor nuclease(R) showed asymmetric distributions and levels of heteroplasmic mutations across mtDNA but no patterns that statistically distinguished sPD from CTL. sPD brain mitochondria displayed reductions of nine respirasome proteins (respiratory complexes I-V). Reduced levels of sPD brain mitochondrial complex II, III and V, but not complex I or IV proteins, correlated closely with rates of NADH-driven electron flow. mtDNA levels and PGC-1alpha expression did not differ between sPD and CTL brains.
PD brain mitochondria have reduced mitochondrial respiratory protein levels in complexes I-V, implying a generalized defect in respirasome assembly. These deficiencies do not appear to arise from altered point mutational burden in mtDNA or reduction of nuclear signaling for mitochondrial biogenesis, implying downstream etiologies. The origin of age-related increases in distribution of oxidative mtDNA damage in sPD but not CTL brains is not clear, tracks with but does not determine the sPD phenotype, and may indicate a unique consequence of aging present in sPD that could contribute to mtDNA deletion generation in addition to mtDNA replication, transcription and sequencing errors. sPD frontal cortex experiences a generalized bioenergetic deficiency above and beyond aging that could contribute to mood disorders and cognitive impairments.
散发性帕金森病(sPD)是一种神经系统疾病,表现为运动迟缓,并常伴有抑郁和认知障碍。sPD 可能有多种相互作用的原因,包括线粒体成分的氧化应激损伤增加和线粒体生物能容量降低。我们分析了 sPD 和 CTL 大脑死后的线粒体,以寻找线粒体 DNA(mtDNA)氧化损伤、异质 mtDNA 点突变和电子传递链蛋白水平的证据。我们试图确定 sPD 大脑是否存在任何 mtDNA 基因型-呼吸表型关系。
用线粒体碱基切除修复酶 8-氧鸟嘌呤糖苷酶-1(hOGG1)处理 sPD 大脑 mtDNA,以年龄依赖性方式抑制了重叠~2 kbase 产物的 qPCR 扩增;CTL 大脑 mtDNA 的扩增对 hOGG1 表现出中等敏感性,不依赖于供体年龄。sPD 和 CTL 大脑中的 hOGG1 mRNA 表达没有差异。使用 Surveyor 核酸内切酶(R)对脑 mtDNA 进行异质体分析显示,mtDNA 中的异质体突变呈不对称分布和水平,但没有模式可以从统计学上区分 sPD 和 CTL。sPD 大脑线粒体显示九个呼吸体蛋白(呼吸复合物 I-V)减少。sPD 大脑线粒体复合物 II、III 和 V 的水平降低,但复合物 I 或 IV 蛋白没有降低,与 NADH 驱动的电子流速率密切相关。sPD 和 CTL 大脑中的 mtDNA 水平和 PGC-1alpha 表达没有差异。
PD 大脑线粒体中复合物 I-V 的呼吸蛋白水平降低,暗示呼吸体组装存在普遍缺陷。这些缺陷似乎不是由于 mtDNA 中突变负担的改变或核信号转导减少引起的线粒体生物发生,暗示了下游病因。sPD 大脑中氧化 mtDNA 损伤分布随年龄增加的原因尚不清楚,但与 sPD 表型有关,但不能决定 sPD 表型,并且可能表明 sPD 中存在独特的衰老后果,除了 mtDNA 复制、转录和测序错误外,还可能导致 mtDNA 缺失的产生。sPD 额叶皮层经历了一种普遍的能量代谢缺陷,超出了衰老的范围,这可能导致情绪障碍和认知障碍。