Yang Chendong, Sudderth Jessica, Dang Tuyen, Bachoo Robert M, McDonald Jeffrey G, DeBerardinis Ralph J
Department of Pediatrics, McDermott Center for Human Growth and Development, University of Texas Southwestern Medical Center at Dallas, Texas 75390-9063, USA.
Cancer Res. 2009 Oct 15;69(20):7986-93. doi: 10.1158/0008-5472.CAN-09-2266. Epub 2009 Oct 13.
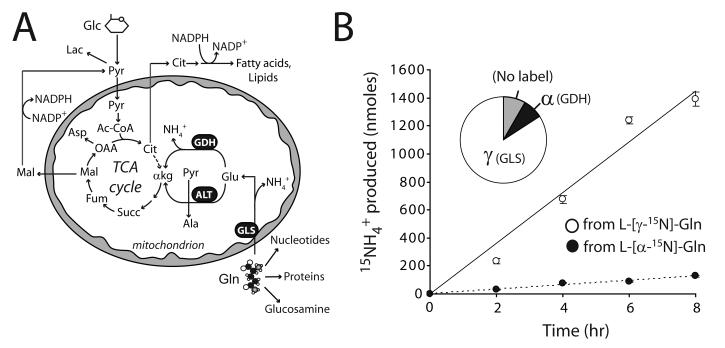
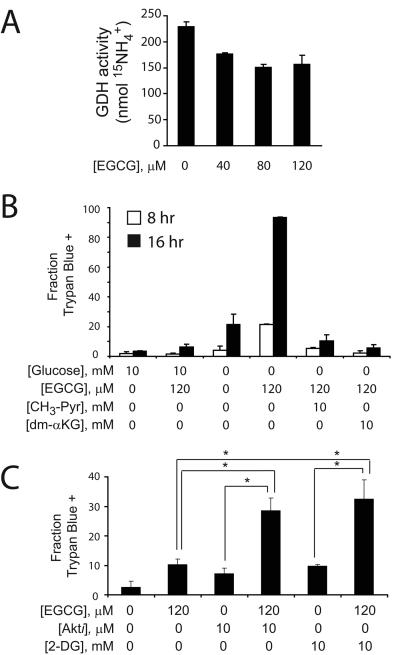
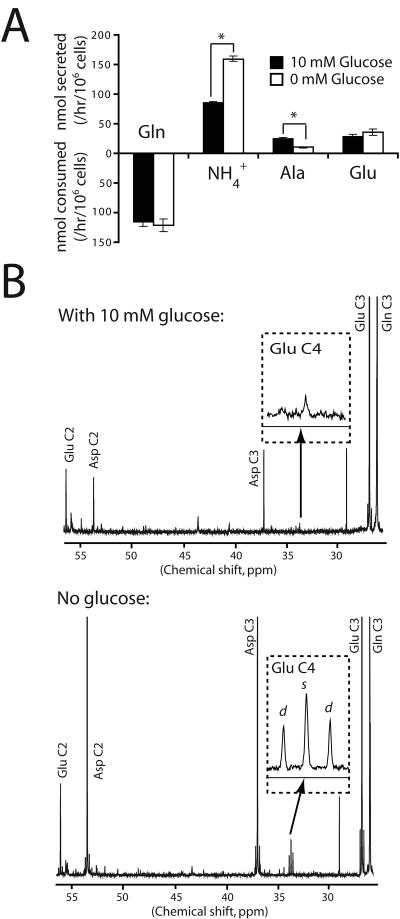
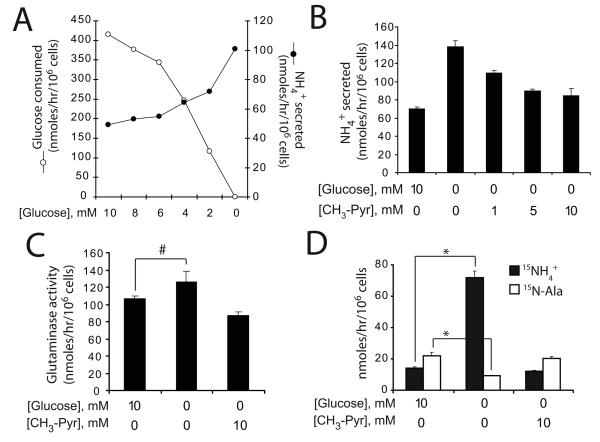
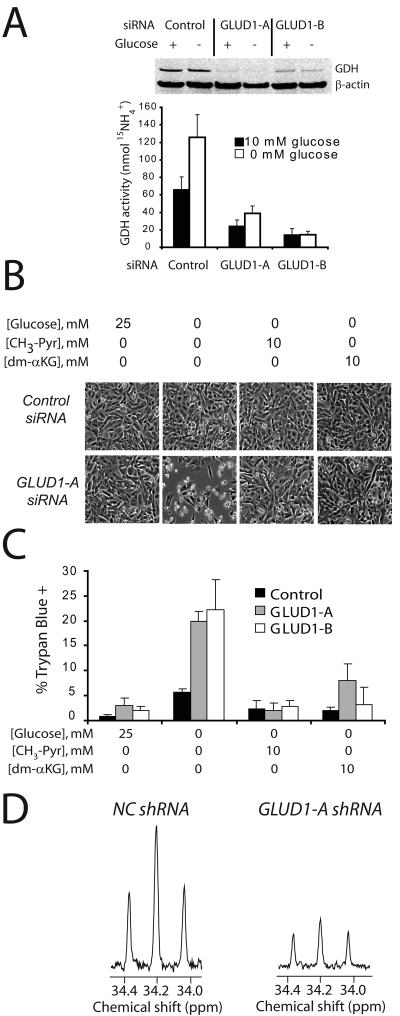
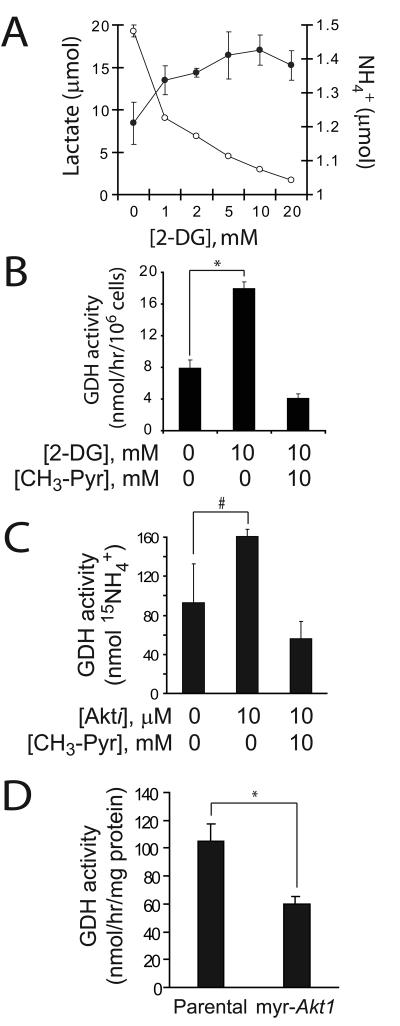
Oncogenes influence nutrient metabolism and nutrient dependence. The oncogene c-Myc stimulates glutamine metabolism and renders cells dependent on glutamine to sustain viability ("glutamine addiction"), suggesting that treatments targeting glutamine metabolism might selectively kill c-Myc-transformed tumor cells. However, many current or proposed cancer therapies interfere with the metabolism of glucose, not glutamine. Here, we studied how c-Myc-transformed cells maintained viability when glucose metabolism was impaired. In SF188 glioblastoma cells, glucose deprivation did not affect net glutamine utilization but elicited a switch in the pathways used to deliver glutamine carbon to the tricarboxylic acid cycle, with a large increase in the activity of glutamate dehydrogenase (GDH). The effect on GDH resulted from the loss of glycolysis because it could be mimicked with the glycolytic inhibitor 2-deoxyglucose and reversed with a pyruvate analogue. Furthermore, inhibition of Akt signaling, which facilitates glycolysis, increased GDH activity whereas overexpression of Akt suppressed it, suggesting that Akt indirectly regulates GDH through its effects on glucose metabolism. Suppression of GDH activity with RNA interference or an inhibitor showed that the enzyme is dispensable in cells able to metabolize glucose but is required for cells to survive impairments of glycolysis brought about by glucose deprivation, 2-deoxyglucose, or Akt inhibition. Thus, inhibition of GDH converted these glutamine-addicted cells to glucose-addicted cells. The findings emphasize the integration of glucose metabolism, glutamine metabolism, and oncogenic signaling in glioblastoma cells and suggest that exploiting compensatory pathways of glutamine metabolism can improve the efficacy of cancer treatments that impair glucose utilization.
癌基因影响营养物质代谢和营养物质依赖性。癌基因c-Myc刺激谷氨酰胺代谢,使细胞依赖谷氨酰胺来维持生存能力(“谷氨酰胺成瘾”),这表明针对谷氨酰胺代谢的治疗可能会选择性地杀死c-Myc转化的肿瘤细胞。然而,目前许多现有的或提议的癌症治疗方法干扰的是葡萄糖代谢,而非谷氨酰胺代谢。在此,我们研究了c-Myc转化的细胞在葡萄糖代谢受损时是如何维持生存能力的。在SF188胶质母细胞瘤细胞中,葡萄糖剥夺并不影响谷氨酰胺的净利用,但引发了用于将谷氨酰胺碳输送到三羧酸循环的途径的转变,谷氨酸脱氢酶(GDH)的活性大幅增加。对GDH的影响是由于糖酵解的丧失,因为它可以被糖酵解抑制剂2-脱氧葡萄糖模拟,并被丙酮酸类似物逆转。此外,促进糖酵解的Akt信号通路的抑制增加了GDH活性,而Akt的过表达则抑制了GDH活性,这表明Akt通过其对葡萄糖代谢的影响间接调节GDH。用RNA干扰或抑制剂抑制GDH活性表明,该酶在能够代谢葡萄糖的细胞中是可有可无的,但对于因葡萄糖剥夺、2-脱氧葡萄糖或Akt抑制导致糖酵解受损的细胞的存活是必需的。因此,抑制GDH将这些谷氨酰胺成瘾的细胞转化为葡萄糖成瘾的细胞。这些发现强调了胶质母细胞瘤细胞中葡萄糖代谢、谷氨酰胺代谢和致癌信号通路的整合,并表明利用谷氨酰胺代谢的补偿途径可以提高损害葡萄糖利用的癌症治疗的疗效。