University of Copenhagen, Department of Computer Science, Universitetsparken 1, 2100 Copenhagen, Denmark.
BMC Bioinformatics. 2009 Oct 16;10:338. doi: 10.1186/1471-2105-10-338.
Predicting the three-dimensional structure of a protein from its amino acid sequence is currently one of the most challenging problems in bioinformatics. The internal structure of helices and sheets is highly recurrent and help reduce the search space significantly. However, random coil segments make up nearly 40% of proteins and they do not have any apparent recurrent patterns, which complicates overall prediction accuracy of protein structure prediction methods. Luckily, previous work has indicated that coil segments are in fact not completely random in structure and flanking residues do seem to have a significant influence on the dihedral angles adopted by the individual amino acids in coil segments. In this work we attempt to predict a probability distribution of these dihedral angles based on the flanking residues. While attempts to predict dihedral angles of coil segments have been done previously, none have, to our knowledge, presented comparable results for the probability distribution of dihedral angles.
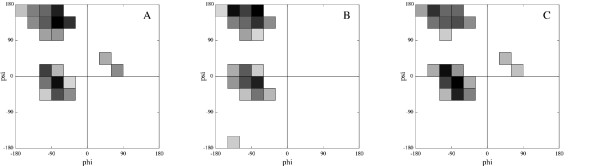
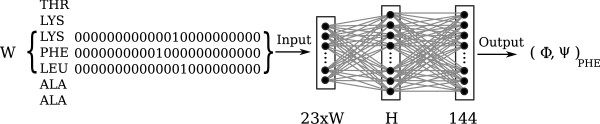
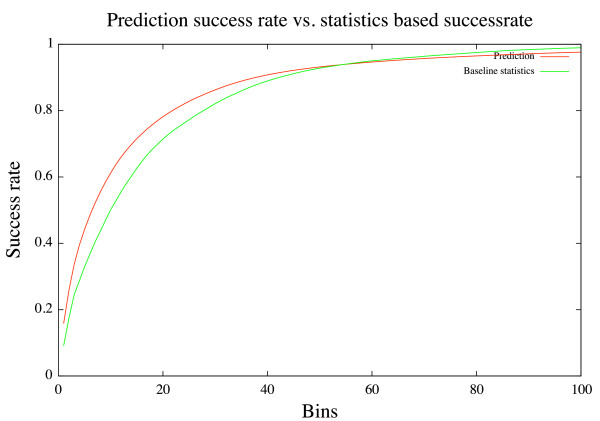
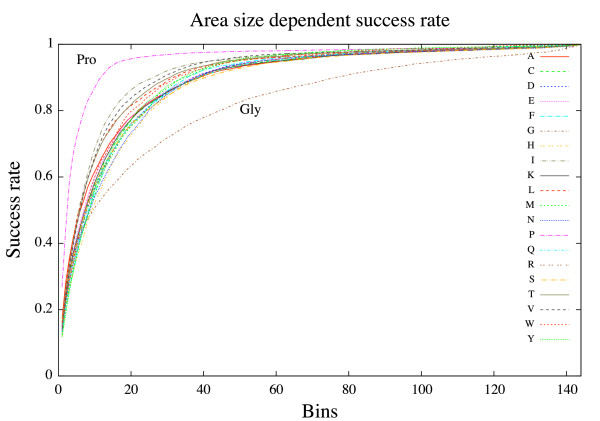
In this paper we develop an artificial neural network that uses an input-window of amino acids to predict a dihedral angle probability distribution for the middle residue in the input-window. The trained neural network shows a significant improvement (4-68%) in predicting the most probable bin (covering a 30 degrees x 30 degrees area of the dihedral angle space) for all amino acids in the data set compared to baseline statistics. An accuracy comparable to that of secondary structure prediction ( approximately 80%) is achieved by observing the 20 bins with highest output values.
Many different protein structure prediction methods exist and each uses different tools and auxiliary predictions to help determine the native structure. In this work the sequence is used to predict local context dependent dihedral angle propensities in coil-regions. This predicted distribution can potentially improve tertiary structure prediction methods that are based on sampling the backbone dihedral angles of individual amino acids. The predicted distribution may also help predict local structure fragments used in fragment assembly methods.
从氨基酸序列预测蛋白质的三维结构是目前生物信息学中最具挑战性的问题之一。螺旋和片层的内部结构高度重复,这有助于显著缩小搜索空间。然而,无规卷曲片段约占蛋白质的 40%,它们没有任何明显的重复模式,这使得蛋白质结构预测方法的整体预测准确性变得复杂。幸运的是,以前的工作表明,卷曲片段实际上在结构上并不是完全随机的,并且侧翼残基似乎对卷曲片段中各个氨基酸所采用的二面角有显著影响。在这项工作中,我们试图根据侧翼残基预测这些二面角的概率分布。虽然以前已经尝试预测卷曲片段的二面角,但据我们所知,没有一个方法能够为二面角的概率分布提供可比的结果。
在本文中,我们开发了一个人工神经网络,该网络使用氨基酸输入窗口来预测输入窗口中间残基的二面角概率分布。与基线统计相比,训练有素的神经网络在预测数据集所有氨基酸的最可能-bin(覆盖二面角空间的 30 度 x 30 度区域)方面显示出显著的改进(4-68%)。通过观察输出值最高的 20 个-bin,可以达到与二级结构预测相当的精度(约 80%)。
存在许多不同的蛋白质结构预测方法,每种方法都使用不同的工具和辅助预测来帮助确定天然结构。在这项工作中,序列用于预测卷曲区域中局部上下文相关的二面角倾向。这种预测的分布可能有助于改进基于采样单个氨基酸的骨架二面角的三级结构预测方法。预测的分布也可能有助于预测用于片段组装方法的局部结构片段。