Fralin Life Science Institute, Department of Entomology, Virginia Tech, Blacksburg, Virginia, United States of America.
PLoS One. 2009 Oct 22;4(10):e7468. doi: 10.1371/journal.pone.0007468.
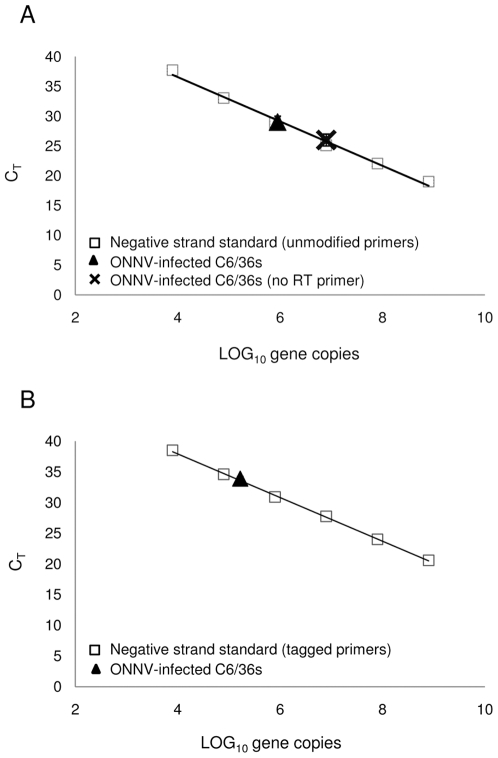
The presence of full-length complements of viral genomic RNA is a hallmark of RNA virus replication within an infected cell. As such, methods for detecting and measuring specific strands of viral RNA in infected cells and tissues are important in the study of RNA viruses. Strand-specific quantitative real-time PCR (ssqPCR) assays are increasingly being used for this purpose, but the accuracy of these assays depends on the assumption that the amount of cDNA measured during the quantitative PCR (qPCR) step accurately reflects amounts of a specific viral RNA strand present in the RT reaction. To specifically test this assumption, we developed multiple ssqPCR assays for the positive-strand RNA virus o'nyong-nyong (ONNV) that were based upon the most prevalent ssqPCR assay design types in the literature. We then compared various parameters of the ONNV-specific assays. We found that an assay employing standard unmodified virus-specific primers failed to discern the difference between cDNAs generated from virus specific primers and those generated through false priming. Further, we were unable to accurately measure levels of ONNV (-) strand RNA with this assay when higher levels of cDNA generated from the (+) strand were present. Taken together, these results suggest that assays of this type do not accurately quantify levels of the anti-genomic strand present during RNA virus infectious cycles. However, an assay permitting the use of a tag-specific primer was able to distinguish cDNAs transcribed from ONNV (-) strand RNA from other cDNAs present, thus allowing accurate quantification of the anti-genomic strand. We also report the sensitivities of two different detection strategies and chemistries, SYBR(R) Green and DNA hydrolysis probes, used with our tagged ONNV-specific ssqPCR assays. Finally, we describe development, design and validation of ssqPCR assays for chikungunya virus (CHIKV), the recent cause of large outbreaks of disease in the Indian Ocean region.
全长互补的病毒基因组 RNA 的存在是病毒在感染细胞内复制的一个标志。因此,检测和测量感染细胞和组织中特定病毒 RNA 链的方法在 RNA 病毒研究中非常重要。用于此目的的越来越多的方法是基于特定链的定量实时 PCR(ssqPCR)检测,但是这些检测的准确性取决于在定量 PCR(qPCR)步骤中测量的 cDNA 量准确反映 RT 反应中存在的特定病毒 RNA 链的数量的假设。为了专门测试此假设,我们开发了针对正链 RNA 病毒 o'nyong-nyong(ONNV)的多个 ssqPCR 检测方法,这些检测方法基于文献中最常见的 ssqPCR 检测设计类型。然后,我们比较了 ONNV 特异性检测方法的各种参数。我们发现,使用标准未修饰的病毒特异性引物的检测方法无法区分病毒特异性引物产生的 cDNA 与通过错误引发产生的 cDNA 之间的差异。此外,当存在更高水平的来自(+)链的 cDNA 时,我们无法使用该检测准确测量 ONNV(-)链 RNA 的水平。总的来说,这些结果表明,此类检测方法不能准确地定量 RNA 病毒感染周期中存在的反基因组链的水平。但是,允许使用标记特异性引物的检测方法能够区分从 ONNV(-)链 RNA 转录的 cDNA 与其他存在的 cDNA,从而能够准确地定量反基因组链。我们还报告了两种不同的检测策略和化学方法(SYBR(R)Green 和 DNA 水解探针)的灵敏度,这些方法与我们标记的 ONNV 特异性 ssqPCR 检测方法一起使用。最后,我们描述了寨卡病毒(CHIKV)的 ssqPCR 检测方法的开发,设计和验证,寨卡病毒是印度洋地区最近大规模爆发疾病的原因。