McDonald Sarah M, Matthijnssens Jelle, McAllen John K, Hine Erin, Overton Larry, Wang Shiliang, Lemey Philippe, Zeller Mark, Van Ranst Marc, Spiro David J, Patton John T
Laboratory of Infectious Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland, USA.
PLoS Pathog. 2009 Oct;5(10):e1000634. doi: 10.1371/journal.ppat.1000634. Epub 2009 Oct 23.
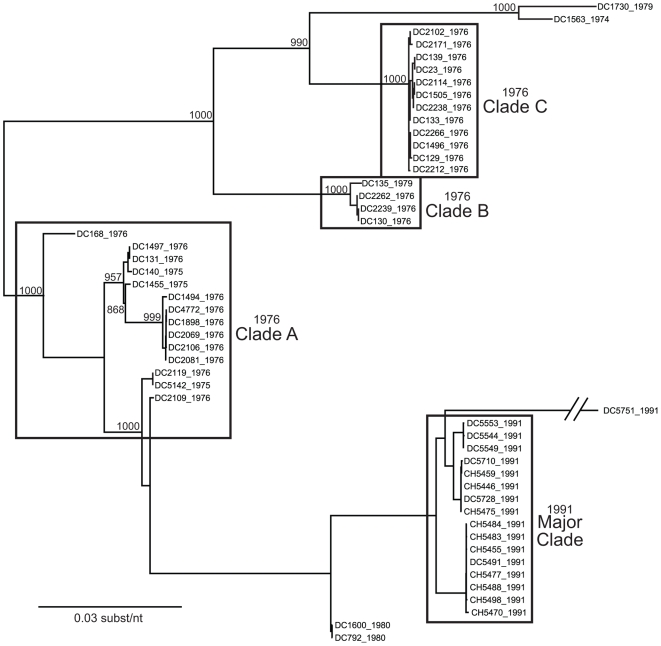
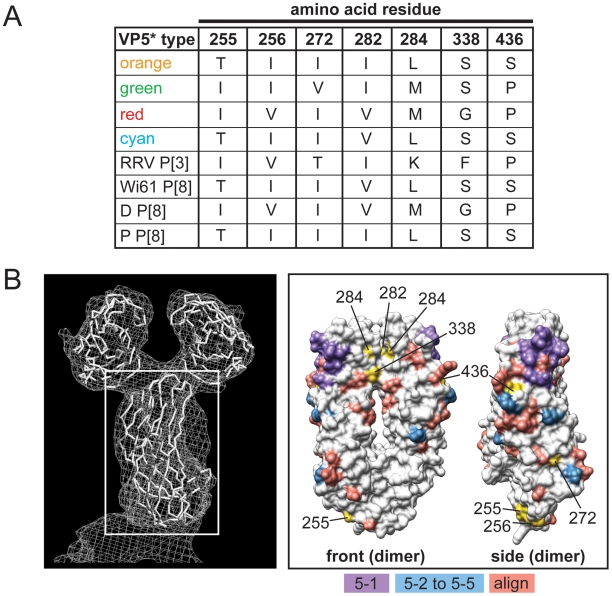
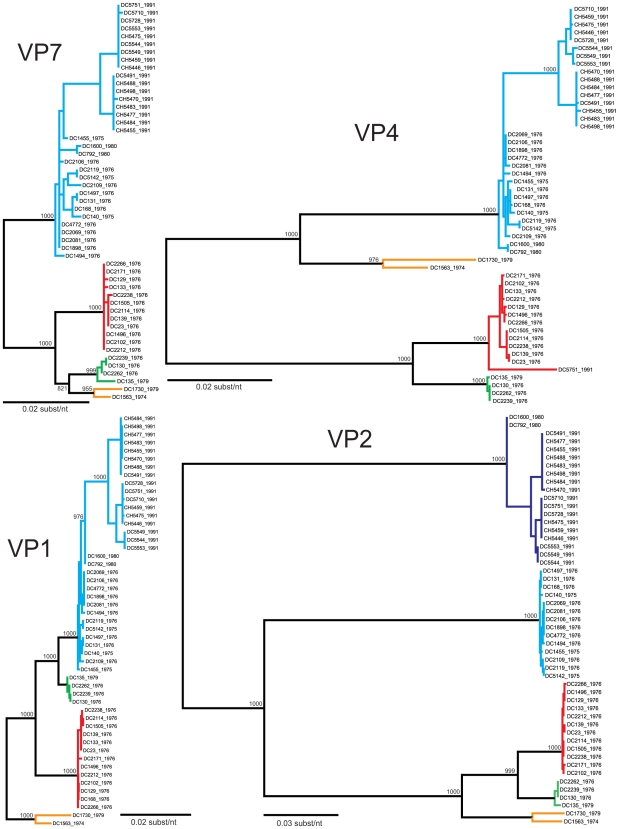
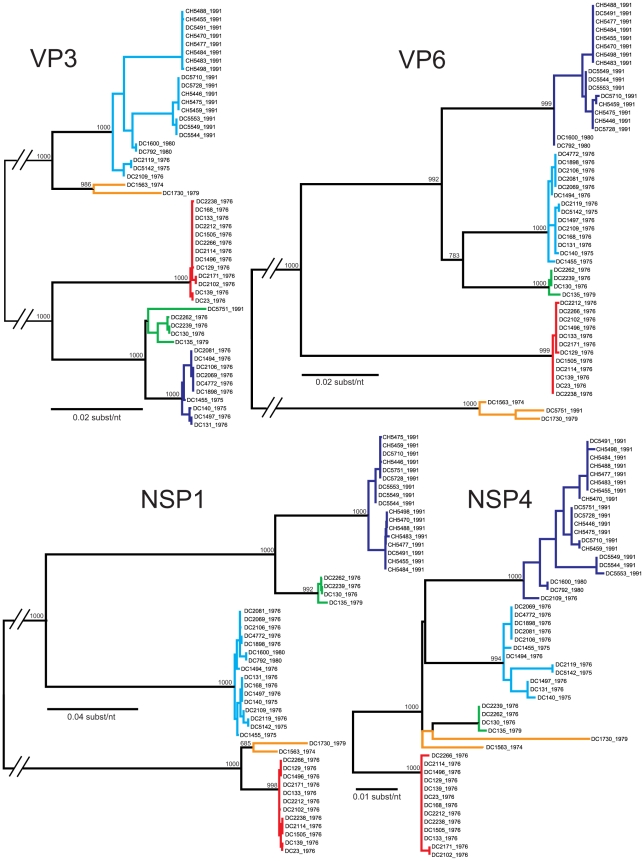
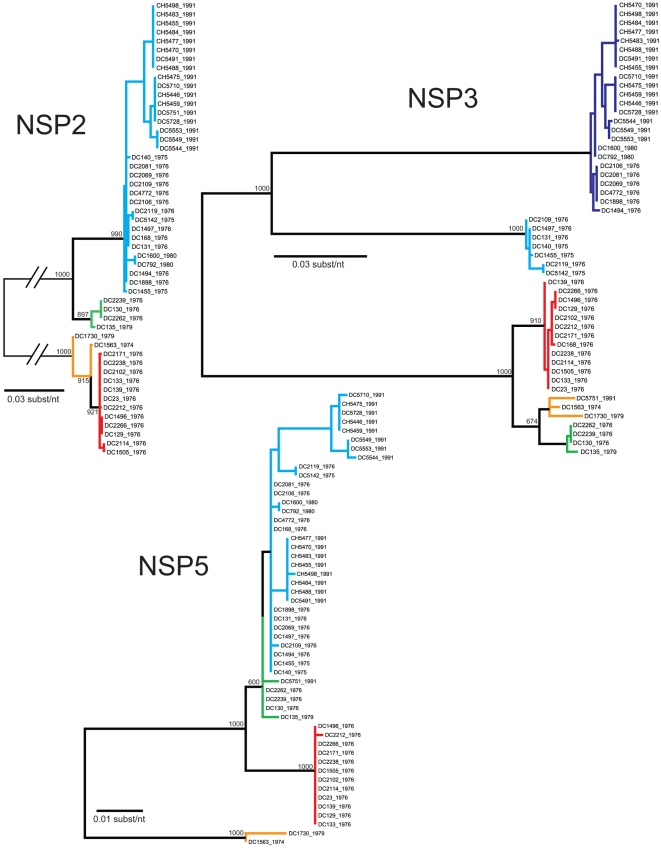
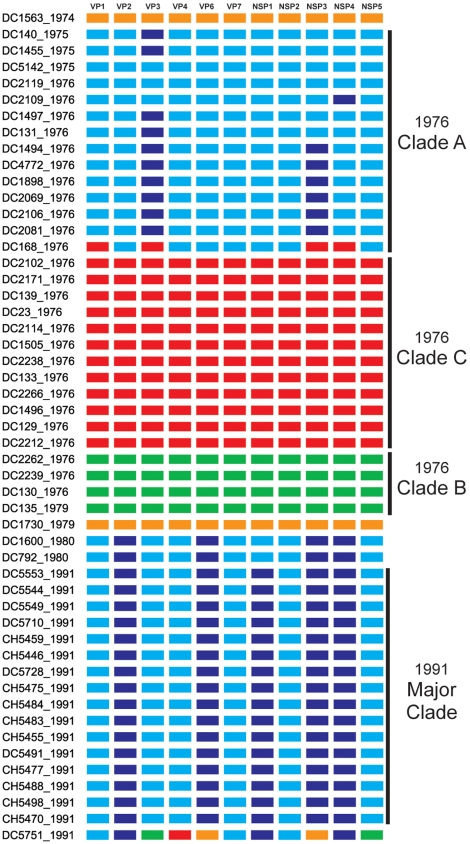
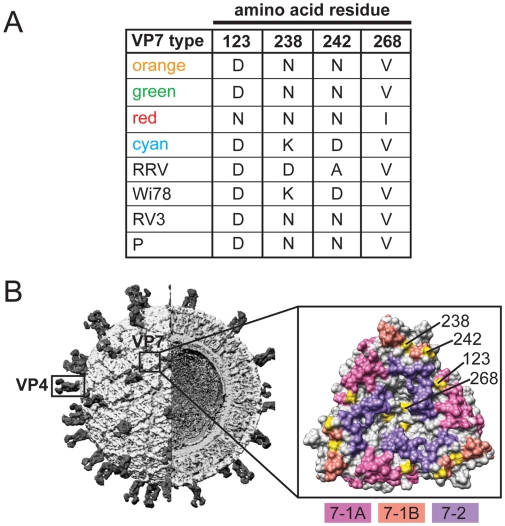
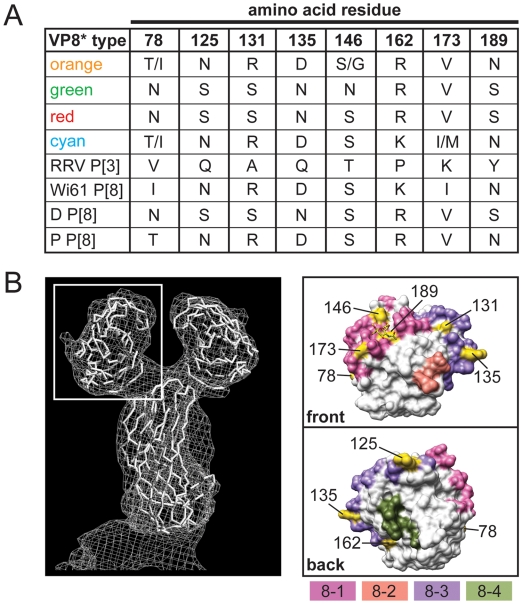
Group A human rotaviruses (RVs) are a major cause of severe gastroenteritis in infants and young children. Yet, aside from the genes encoding serotype antigens (VP7; G-type and VP4; P-type), little is known about the genetic make-up of emerging and endemic human RV strains. To gain insight into the diversity and evolution of RVs circulating at a single location over a period of time, we sequenced the eleven-segmented, double-stranded RNA genomes of fifty-one G3P[8] strains collected from 1974 to 1991 at Children's Hospital National Medical Center, Washington, D. C. During this period, G1P[8] strains typically dominated, comprising on average 56% of RV infections each year in hospitalized children. A notable exception was in the 1976 and 1991 winter seasons when the incidence of G1P[8] infections decreased dramatically, a trend that correlated with a significant increase in G3P[8] infections. Our sequence analysis indicates that the 1976 season was characterized by the presence of several genetically distinct, co-circulating clades of G3P[8] viruses, which contained minor but significant differences in their encoded proteins. These 1976 lineages did not readily exchange gene segments with each other, but instead remained stable over the course of the season. In contrast, the 1991 season contained a single major clade, whose genome constellation was similar to one of the 1976 clades. The 1991 clade may have gained a fitness advantage after reassorting with as of yet unidentified RV strain(s). This study reveals for the first time that genetically distinct RV clades of the same G/P-type can co-circulate and cause disease. The findings from this study also suggest that, although gene segment exchange occurs, most reassortant strains are replaced over time by lineages with preferred genome constellations. Elucidation of the selective pressures that favor maintenance of RVs with certain sets of genes may be necessary to anticipate future vaccine needs.
A组人类轮状病毒(RVs)是婴幼儿严重肠胃炎的主要病因。然而,除了编码血清型抗原的基因(VP7;G型和VP4;P型)外,对于新出现的和地方性流行的人类RV毒株的基因组成了解甚少。为了深入了解一段时间内在单一地点传播的RVs的多样性和进化情况,我们对1974年至1991年期间在华盛顿特区儿童医院国家医疗中心收集的51株G3P[8]毒株的11节段双链RNA基因组进行了测序。在此期间,G1P[8]毒株通常占主导地位,在住院儿童中每年平均占RV感染的56%。一个显著的例外是在1976年和1991年冬季,G1P[8]感染的发病率急剧下降,这一趋势与G3P[8]感染的显著增加相关。我们的序列分析表明,1976年季节的特点是存在几个基因上不同的、共同传播的G3P[8]病毒分支,它们编码的蛋白质存在微小但显著的差异。这些1976年的谱系彼此之间不容易交换基因片段,而是在整个季节中保持稳定。相比之下,1991年季节包含一个单一的主要分支,其基因组组合与1976年的一个分支相似。1991年的分支可能在与尚未确定的RV毒株重配后获得了适应性优势。这项研究首次揭示了相同G/P型的基因上不同的RV分支可以共同传播并引发疾病。这项研究的结果还表明,尽管发生了基因片段交换,但随着时间的推移,大多数重配毒株会被具有优选基因组组合的谱系所取代。阐明有利于维持具有某些基因组合的RVs的选择压力可能对于预测未来的疫苗需求是必要的。