Department of Zoology, George S Wise Faculty of Life Sciences, Tel-Aviv University, Tel Aviv 69978, Israel.
BMC Genomics. 2009 Nov 17;10:534. doi: 10.1186/1471-2164-10-534.
Tunicates represent a key metazoan group as the sister-group of vertebrates within chordates. The six complete mitochondrial genomes available so far for tunicates have revealed distinctive features. Extensive gene rearrangements and particularly high evolutionary rates have been evidenced with regard to other chordates. This peculiar evolutionary dynamics has hampered the reconstruction of tunicate phylogenetic relationships within chordates based on mitogenomic data.
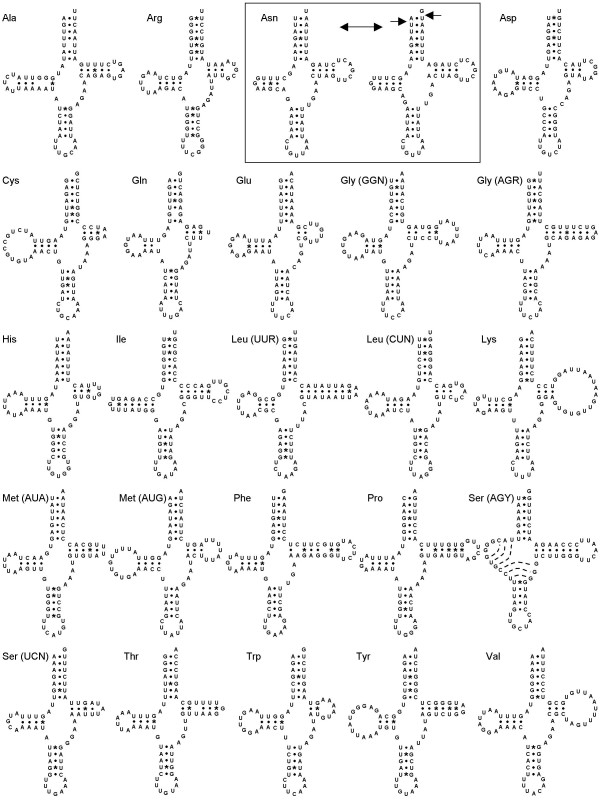
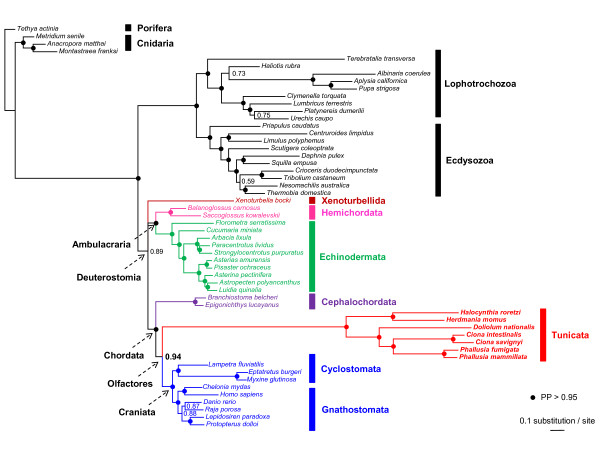
In order to further understand the atypical evolutionary dynamics of the mitochondrial genome of tunicates, we determined the complete sequence of the solitary ascidian Herdmania momus. This genome from a stolidobranch ascidian presents the typical tunicate gene content with 13 protein-coding genes, 2 rRNAs and 24 tRNAs which are all encoded on the same strand. However, it also presents a novel gene arrangement, highlighting the extreme plasticity of gene order observed in tunicate mitochondrial genomes. Probabilistic phylogenetic inferences were conducted on the concatenation of the 13 mitochondrial protein-coding genes from representatives of major metazoan phyla. We show that whereas standard homogeneous amino acid models support an artefactual sister position of tunicates relative to all other bilaterians, the CAT and CAT+BP site- and time-heterogeneous mixture models place tunicates as the sister-group of vertebrates within monophyletic chordates. Moreover, the reference phylogeny indicates that tunicate mitochondrial genomes have experienced a drastic acceleration in their evolutionary rate that equally affects protein-coding and ribosomal-RNA genes.
This is the first mitogenomic study supporting the new chordate phylogeny revealed by recent phylogenomic analyses. It illustrates the beneficial effects of an increased taxon sampling coupled with the use of more realistic amino acid substitution models for the reconstruction of animal phylogeny.
被囊动物是脊索动物中与脊椎动物亲缘关系最近的动物门,代表了一个关键的后生动物群。迄今为止,已获得的 6 个完整的被囊动物线粒体基因组揭示了独特的特征。与其他脊索动物相比,广泛的基因重排和特别高的进化率已经得到证实。这种特殊的进化动态阻碍了基于线粒体基因组数据重建被囊动物在脊索动物中的系统发育关系。
为了进一步了解被囊动物线粒体基因组的非典型进化动态,我们测定了单体被囊动物海鞘的完整序列。这个来自固肠海鞘的基因组具有典型的被囊动物基因含量,包含 13 个蛋白质编码基因、2 个 rRNA 和 24 个 tRNA,它们都编码在同一条链上。然而,它也呈现出一种新的基因排列,突出了被囊动物线粒体基因组中观察到的基因顺序的极端可塑性。对主要后生动物门代表的 13 个线粒体蛋白质编码基因的串联进行了概率系统发育推断。我们表明,虽然标准的同质氨基酸模型支持被囊动物相对于所有其他两侧对称动物的姐妹位置是人为的,但 CAT 和 CAT+BP 位点和时间异质混合模型将被囊动物置于单系脊索动物中的脊椎动物姐妹群。此外,参考系统发育表明,被囊动物的线粒体基因组经历了其进化率的急剧加速,这同样影响了蛋白质编码和核糖体 RNA 基因。
这是第一个支持最近系统基因组分析揭示的新脊索动物系统发育的线粒体基因组研究。它说明了增加分类群采样并结合使用更现实的氨基酸替代模型对动物系统发育进行重建的有益效果。