Department of Biotechnology, University of Verona, Strada le Grazie 15, 37134 Verona, Italy.
BMC Genomics. 2009 Nov 24;10:555. doi: 10.1186/1471-2164-10-555.
The next generation sequencing technologies provide new options to characterize the transcriptome and to develop affordable tools for functional genomics. We describe here an innovative approach for this purpose and demonstrate its potential also for non-model species.
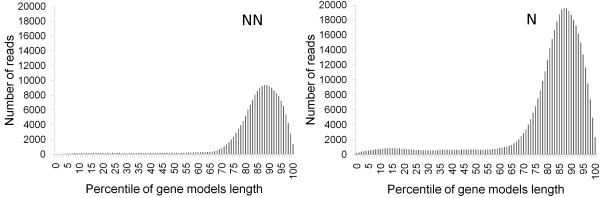
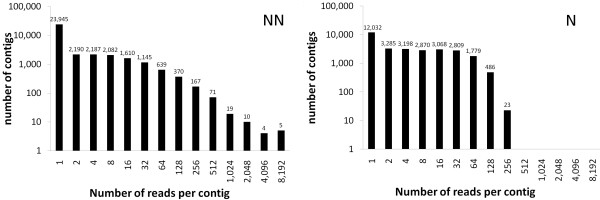
The method we developed is based on 454 sequencing of 3' cDNA fragments from a normalized library constructed from pooled RNAs to generate, through de novo reads assembly, a large catalog of unique transcripts in organisms for which a comprehensive collection of transcripts or the complete genome sequence, is not available. This "virtual transcriptome" provides extensive coverage depth, and can be used for the setting up of a comprehensive microarray based expression analysis. We evaluated the potential of this approach by monitoring gene expression during berry maturation in Vitis vinifera as if no other sequence information was available for this species. The microarray designed on the berries' transcriptome derived from half of a 454 run detected the expression of 19,609 genes, and proved to be more informative than one of the most comprehensive grape microarrays available to date, the GrapeArray 1.2 developed by the Italian-French Public Consortium for Grapevine Genome Characterization, which could detect the expression of 15,556 genes in the same samples.
This approach provides a powerful method to rapidly build up an extensive catalog of unique transcripts that can be successfully used to develop a microarray for large scale analysis of gene expression in any species, without the need for prior sequence knowledge.
下一代测序技术为转录组的特征描述和开发经济实惠的功能基因组学工具提供了新的选择。我们在这里描述了一种用于此目的的创新方法,并证明其对于非模式物种也具有潜力。
我们开发的方法基于 3' cDNA 片段的 454 测序,这些片段来自于从混合 RNA 构建的标准化文库,通过从头读取组装,为那些没有全面转录组或完整基因组序列的生物生成大量独特转录本的目录。这个“虚拟转录组”提供了广泛的覆盖深度,并可用于建立基于微阵列的综合表达分析。我们通过在葡萄浆果成熟过程中监测基因表达来评估这种方法的潜力,就好像该物种没有其他序列信息可用一样。在 454 次运行中设计的一半的葡萄转录组微阵列检测到 19609 个基因的表达,并且证明比迄今为止最全面的葡萄微阵列之一,即由意大利-法国葡萄基因组特征公共联合体开发的 GrapeArray 1.2 更具信息量,后者可以在相同的样本中检测到 15556 个基因的表达。
这种方法提供了一种快速构建大量独特转录本目录的强大方法,可以成功地用于开发微阵列,用于任何物种的大规模基因表达分析,而无需事先了解序列信息。