Sushmita Roy Computer Science, University of New Mexico, Albuquerque, New Mexico, United States of America.
PLoS One. 2009 Nov 20;4(11):e7813. doi: 10.1371/journal.pone.0007813.
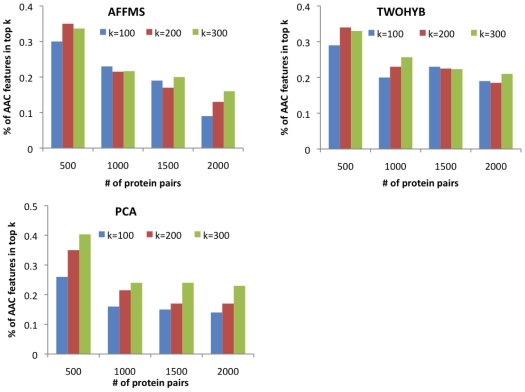
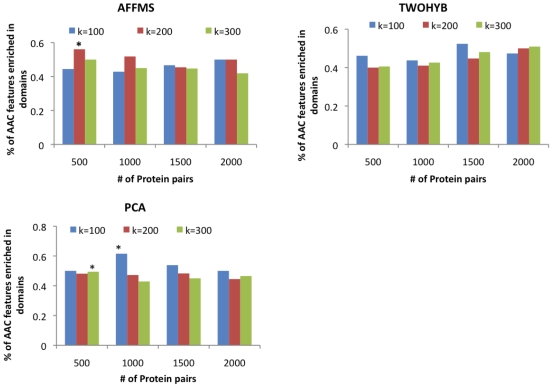
Computational prediction of protein interactions typically use protein domains as classifier features because they capture conserved information of interaction surfaces. However, approaches relying on domains as features cannot be applied to proteins without any domain information. In this paper, we explore the contribution of pure amino acid composition (AAC) for protein interaction prediction. This simple feature, which is based on normalized counts of single or pairs of amino acids, is applicable to proteins from any sequenced organism and can be used to compensate for the lack of domain information.
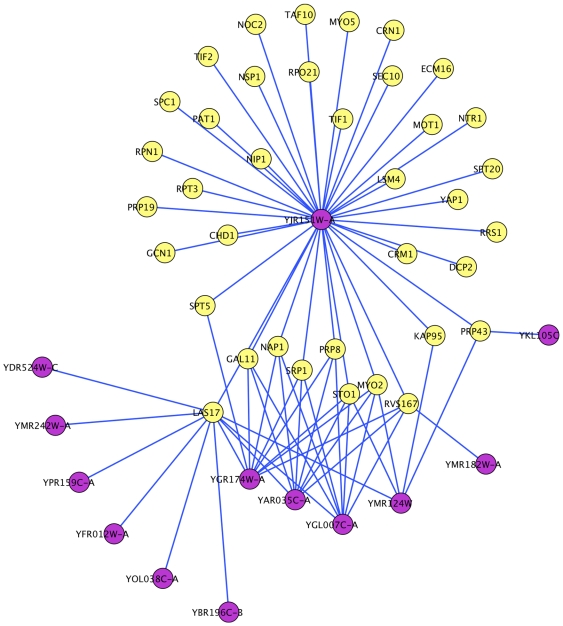
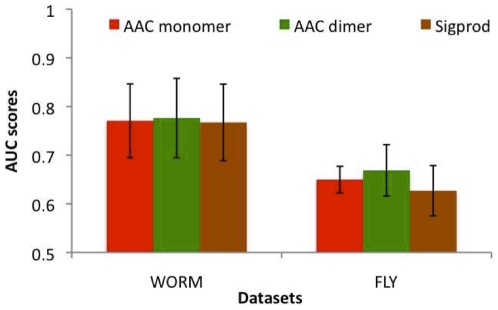
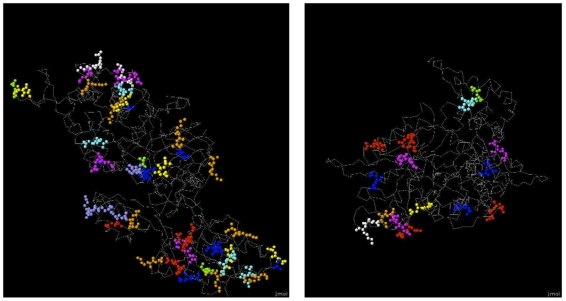
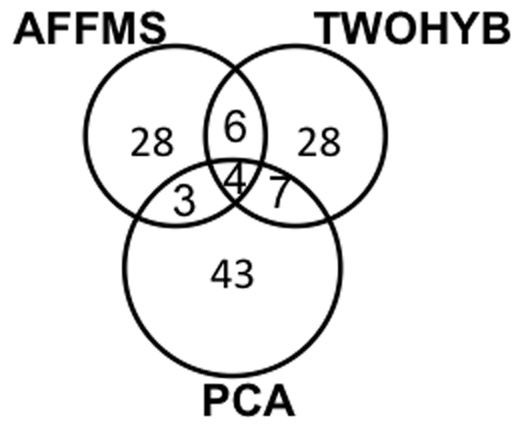
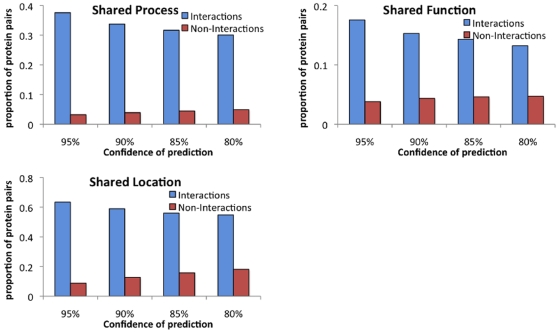
AAC performed at par with protein interaction prediction based on domains on three yeast protein interaction datasets. Similar behavior was obtained using different classifiers, indicating that our results are a function of features and not of classifiers. In addition to yeast datasets, AAC performed comparably on worm and fly datasets. Prediction of interactions for the entire yeast proteome identified a large number of novel interactions, the majority of which co-localized or participated in the same processes. Our high confidence interaction network included both well-studied and uncharacterized proteins. Proteins with known function were involved in actin assembly and cell budding. Uncharacterized proteins interacted with proteins involved in reproduction and cell budding, thus providing putative biological roles for the uncharacterized proteins.
AAC is a simple, yet powerful feature for predicting protein interactions, and can be used alone or in conjunction with protein domains to predict new and validate existing interactions. More importantly, AAC alone performs at par with existing, but more complex, features indicating the presence of sequence-level information that is predictive of interaction, but which is not necessarily restricted to domains.
计算蛋白质相互作用的预测通常使用蛋白质结构域作为分类器特征,因为它们捕获了相互作用表面的保守信息。然而,依赖结构域作为特征的方法不能应用于没有任何结构域信息的蛋白质。在本文中,我们探讨了纯氨基酸组成(AAC)对蛋白质相互作用预测的贡献。这个简单的特征是基于单个或氨基酸对的归一化计数,适用于任何测序生物的蛋白质,可用于弥补结构域信息的缺乏。
AAC 在三个酵母蛋白质相互作用数据集上的表现与基于结构域的蛋白质相互作用预测相当。使用不同的分类器获得了相似的行为,这表明我们的结果是特征的函数,而不是分类器的函数。除了酵母数据集外,AAC 在蠕虫和苍蝇数据集上的表现也相当。对整个酵母蛋白质组的相互作用预测确定了大量新的相互作用,其中大多数共定位或参与相同的过程。我们的高置信度相互作用网络包括了研究充分的和未被描述的蛋白质。具有已知功能的蛋白质参与了肌动蛋白组装和细胞出芽。未被描述的蛋白质与涉及繁殖和细胞出芽的蛋白质相互作用,从而为未被描述的蛋白质提供了潜在的生物学作用。
AAC 是一种简单而强大的预测蛋白质相互作用的特征,可以单独使用或与蛋白质结构域结合使用来预测新的和验证现有的相互作用。更重要的是,仅 AAC 与现有的、但更复杂的特征相当,这表明存在序列水平的信息,这些信息是可预测的相互作用,但不一定局限于结构域。