BMC Bioinformatics. 2009 Dec 2;10:396. doi: 10.1186/1471-2105-10-396.
While substitution matrices can readily be computed from reference alignments, it is challenging to compute optimal or approximately optimal gap penalties. It is also not well understood which substitution matrices are the most effective when alignment accuracy is the goal rather than homolog recognition. Here a new parameter optimization procedure, POP, is described and applied to the problems of optimizing gap penalties and selecting substitution matrices for pair-wise global protein alignments.
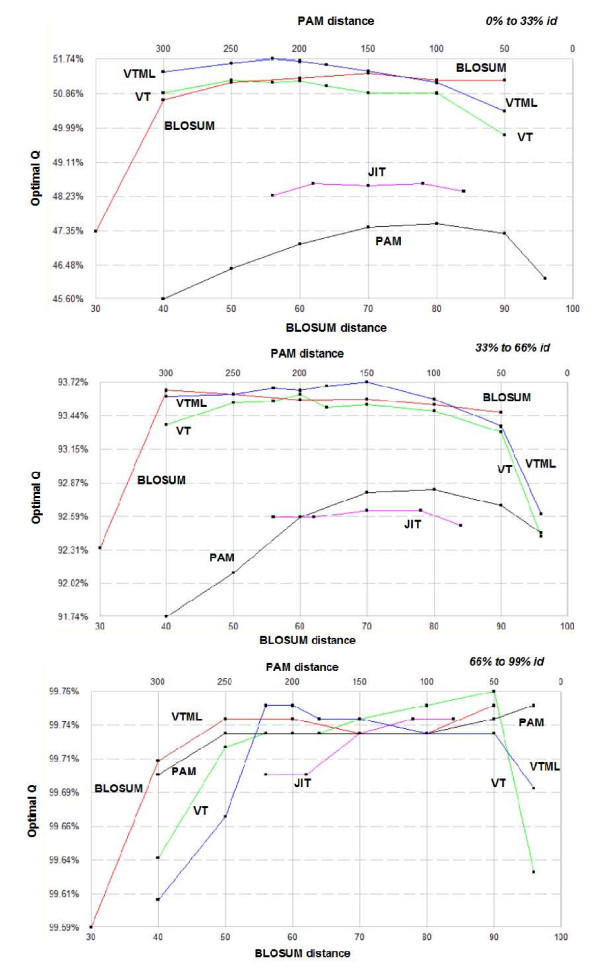
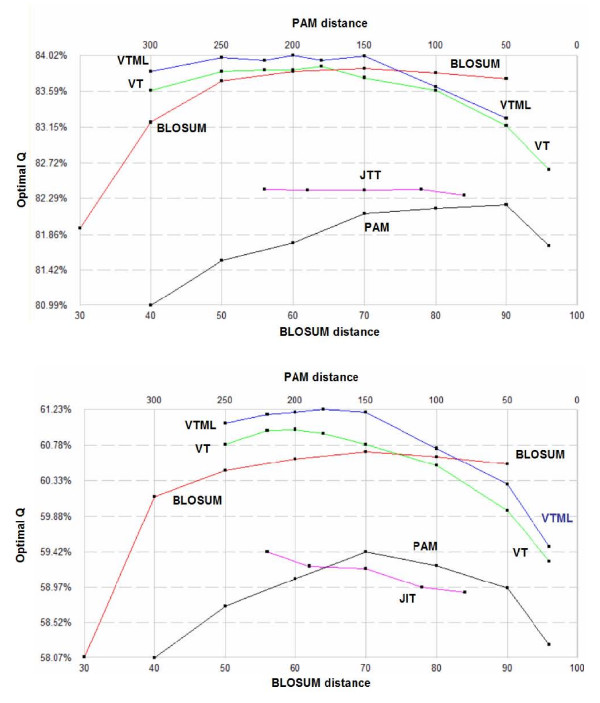
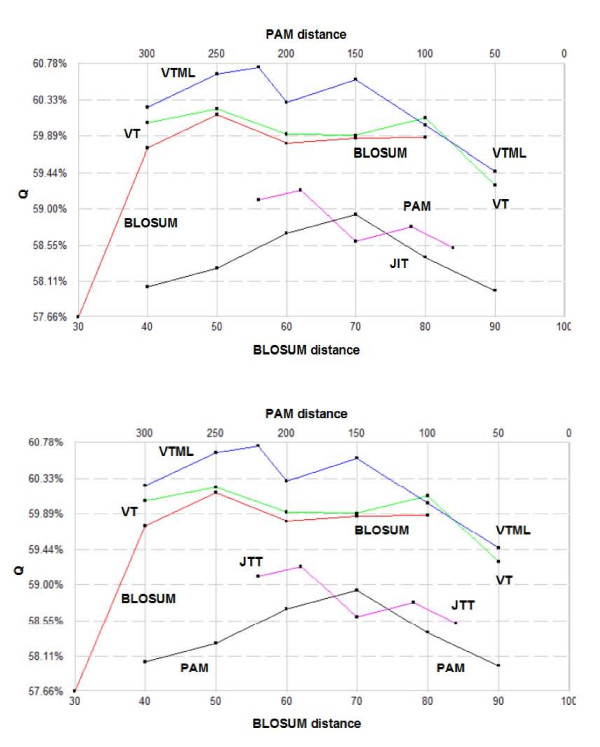
POP is compared to a recent method due to Kim and Kececioglu and found to achieve from 0.2% to 1.3% higher accuracies on pair-wise benchmarks extracted from BALIBASE. The VTML matrix series is shown to be the most accurate on several global pair-wise alignment benchmarks, with VTML200 giving best or close to the best performance in all tests. BLOSUM matrices are found to be slightly inferior, even with the marginal improvements in the bug-fixed RBLOSUM series. The PAM series is significantly worse, giving accuracies typically 2% less than VTML. Integer rounding is found to cause slight degradations in accuracy. No evidence is found that selecting a matrix based on sequence divergence improves accuracy, suggesting that the use of this heuristic in CLUSTALW may be ineffective. Using VTML200 is found to improve the accuracy of CLUSTALW by 8% on BALIBASE and 5% on PREFAB.
The hypothesis that more accurate alignments of distantly related sequences may be achieved using low-identity matrices is shown to be false for commonly used matrix types. Source code and test data is freely available from the author's web site at http://www.drive5.com/pop.
虽然替换矩阵可以从参考比对中轻松计算出来,但计算最佳或近似最佳的空位罚分是具有挑战性的。当目标是对齐准确性而不是同源识别时,哪种替换矩阵最有效也不太清楚。这里描述了一种新的参数优化程序 POP,并将其应用于优化空位罚分和选择用于两两全局蛋白质比对的替换矩阵的问题。
POP 与 Kim 和 Kececioglu 的最新方法进行了比较,在从 BALIBASE 中提取的两两基准测试中,POP 实现了 0.2%至 1.3%的更高准确性。在几个全局两两比对基准测试中,VTML 矩阵系列被证明是最准确的,VTML200 在所有测试中都表现出最佳或接近最佳的性能。BLOSUM 矩阵稍逊一筹,即使在修正后的 RBLOSUM 系列中略有改进。PAM 系列明显较差,准确性通常比 VTML 低 2%。发现整数舍入会导致准确性略有下降。没有证据表明根据序列分歧选择矩阵会提高准确性,这表明 CLUSTALW 中使用此启发式可能无效。在 BALIBASE 上,使用 VTML200 可将 CLUSTALW 的准确性提高 8%,在 PREFAB 上可提高 5%。
对于常用的矩阵类型,远缘序列的更准确比对可能使用低同一性矩阵来实现的假设被证明是错误的。源代码和测试数据可从作者的网站(http://www.drive5.com/pop)免费获得。