Keul Frank, Hess Martin, Goesele Michael, Hamacher Kay
Computational Biology and Simulation, Department of Biology, Technische Universität Darmstadt, Schnittspahnstraße 2, Darmstadt, 64287, Germany.
Graphics, Capture and Massively Parallel Computing, Department of Computer Science, Technische Universität Darmstadt, Rundeturmstraße 12, Darmstadt, 64283, Germany.
BMC Bioinformatics. 2017 Jun 5;18(1):293. doi: 10.1186/s12859-017-1703-z.
Detecting homologous protein sequences and computing multiple sequence alignments (MSA) are fundamental tasks in molecular bioinformatics. These tasks usually require a substitution matrix for modeling evolutionary substitution events derived from a set of aligned sequences. Over the last years, the known sequence space increased drastically and several publications demonstrated that this can lead to significantly better performing matrices. Interestingly, matrices based on dated sequence datasets are still the de facto standard for both tasks even though their data basis may limit their capabilities. We address these aspects by presenting a new substitution matrix series called PFASUM. These matrices are derived from Pfam seed MSAs using a novel algorithm and thus build upon expert ground truth data covering a large and diverse sequence space.
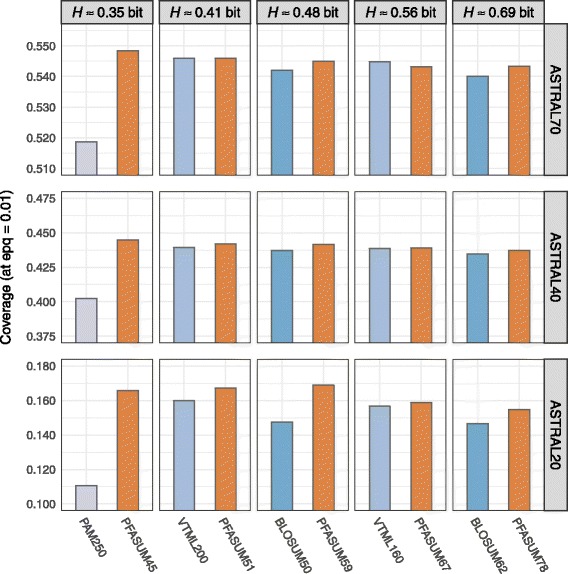
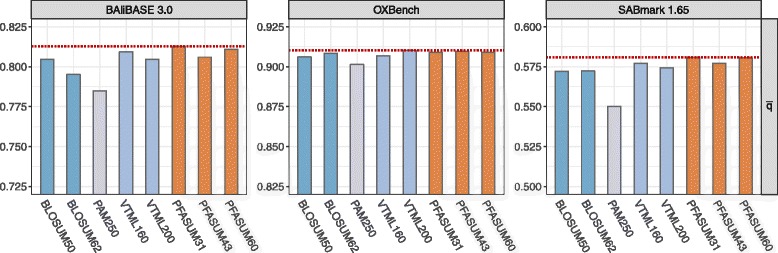
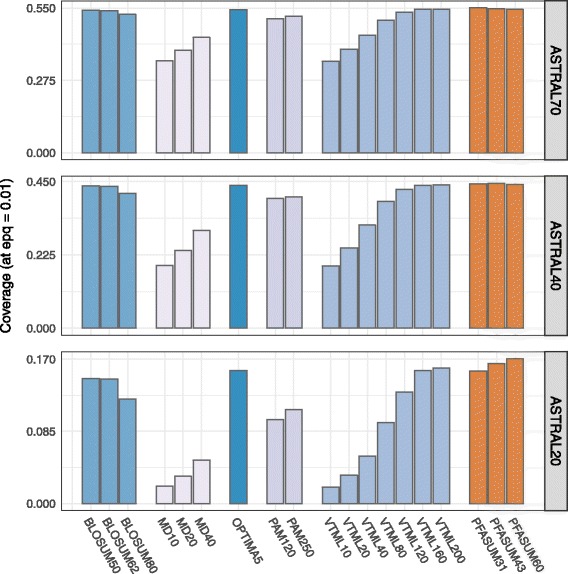
We show results for two use cases: First, we tested the homology search performance of PFASUM matrices on up-to-date ASTRAL databases with varying sequence similarity. Our study shows that the usage of PFASUM matrices can lead to significantly better homology search results when compared to conventional matrices. PFASUM matrices with comparable relative entropies to the commonly used substitution matrices BLOSUM50, BLOSUM62, PAM250, VTML160 and VTML200 outperformed their corresponding counterparts in 93% of all test cases. A general assessment also comparing matrices with different relative entropies showed that PFASUM matrices delivered the best homology search performance in the test set. Second, our results demonstrate that the usage of PFASUM matrices for MSA construction improves their quality when compared to conventional matrices. On up-to-date MSA benchmarks, at least 60% of all MSAs were reconstructed in an equal or higher quality when using MUSCLE with PFASUM31, PFASUM43 and PFASUM60 matrices instead of conventional matrices. This rate even increases to at least 76% for MSAs containing similar sequences.
We present the novel PFASUM substitution matrices derived from manually curated MSA ground truth data covering the currently known sequence space. Our results imply that PFASUM matrices improve homology search performance as well as MSA quality in many cases when compared to conventional substitution matrices. Hence, we encourage the usage of PFASUM matrices and especially PFASUM60 for these specific tasks.
检测同源蛋白质序列和计算多序列比对(MSA)是分子生物信息学中的基本任务。这些任务通常需要一个替换矩阵来模拟从一组比对序列中得出的进化替换事件。在过去几年中,已知的序列空间急剧增加,一些出版物表明这可以导致性能显著更好的矩阵。有趣的是,基于过时序列数据集的矩阵仍然是这两项任务的事实上的标准,尽管它们的数据基础可能会限制其能力。我们通过提出一个名为PFASUM的新替换矩阵系列来解决这些问题。这些矩阵是使用一种新颖的算法从Pfam种子MSA中推导出来的,因此建立在覆盖广泛且多样的序列空间的专家真值数据之上。
我们展示了两个用例的结果:第一,我们在具有不同序列相似性的最新ASTRAL数据库上测试了PFASUM矩阵的同源性搜索性能。我们的研究表明,与传统矩阵相比,使用PFASUM矩阵可以显著提高同源性搜索结果。与常用替换矩阵BLOSUM50、BLOSUM62、PAM250、VTML160和VTML200具有可比相对熵的PFASUM矩阵在所有测试案例的93%中优于其相应的对应矩阵。一项比较不同相对熵矩阵的综合评估还表明,PFASUM矩阵在测试集中提供了最佳的同源性搜索性能。第二,我们的结果表明,与传统矩阵相比,使用PFASUM矩阵进行MSA构建可提高其质量。在最新的MSA基准测试中,当使用带有PFASUM31、PFASUM43和PFASUM60矩阵的MUSCLE而不是传统矩阵时,所有MSA中至少60%被重建为同等或更高质量。对于包含相似序列的MSA,这一比例甚至增加到至少76%。
我们提出了从手动策划的MSA真值数据推导而来的新颖的PFASUM替换矩阵,该数据覆盖了当前已知的序列空间。我们的结果表明,与传统替换矩阵相比,PFASUM矩阵在许多情况下提高了同源性搜索性能以及MSA质量。因此,我们鼓励在这些特定任务中使用PFASUM矩阵,尤其是PFASUM60。