Genome Informatics, Faculty of Technology, Bielefeld University, Bielefeld, Germany.
Bioinformatics. 2010 Feb 15;26(4):570-1. doi: 10.1093/bioinformatics/btp690. Epub 2009 Dec 16.
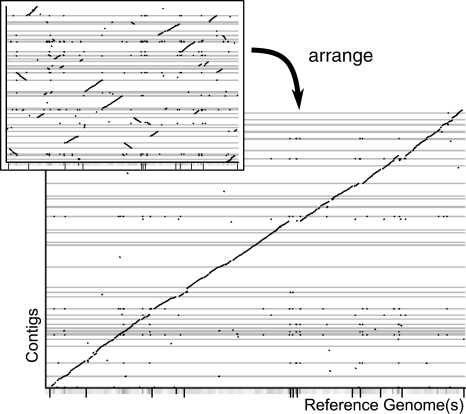
Recent parallel pyrosequencing methods and the increasing number of finished genomes encourage the sequencing and investigation of closely related strains. Although the sequencing itself becomes easier and cheaper with each machine generation, the finishing of the genomes remains difficult. Instead of the desired whole genomic sequence, a set of contigs is the result of the assembly. In this applications note, we present the tool r2cat (related reference contig arrangement tool) that helps in the task of comparative assembly and also provides an interactive visualization for synteny inspection.
最近的平行焦磷酸测序方法和越来越多的完成基因组促使人们对密切相关的菌株进行测序和研究。尽管随着每一代机器的发展,测序本身变得更加容易和廉价,但基因组的完成仍然很困难。组装的结果不是期望的全基因组序列,而是一组 contigs。在本应用说明中,我们介绍了 r2cat(相关参考 contig 排列工具),它有助于比较组装任务,并提供用于检查同线性的交互式可视化。