Institute of Microbiology, ETH Zürich, Zürich, Switzerland.
PLoS Pathog. 2010 Jan;6(1):e1000711. doi: 10.1371/journal.ppat.1000711. Epub 2010 Jan 8.
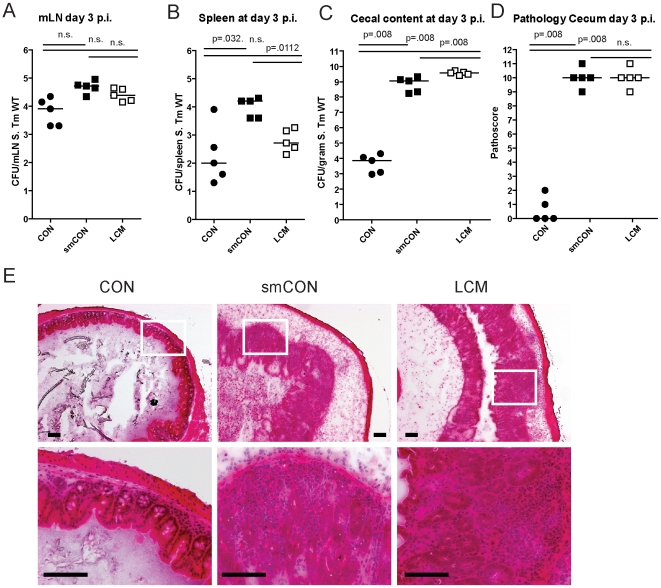
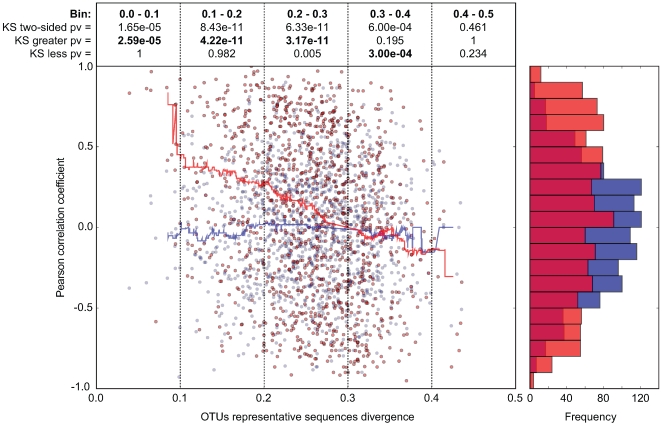
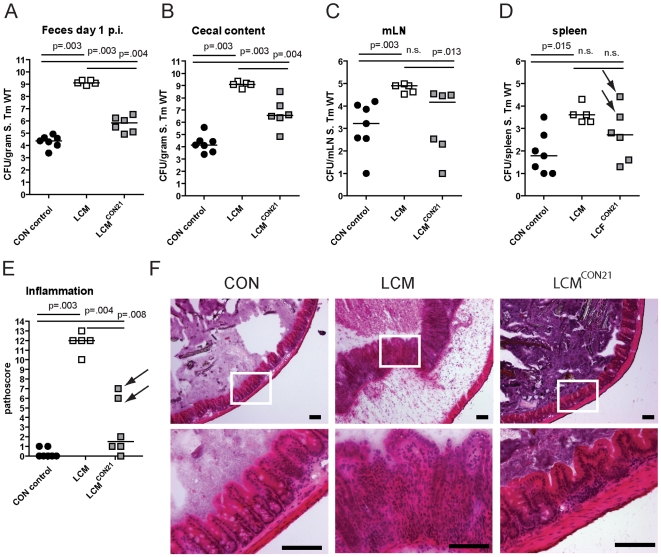
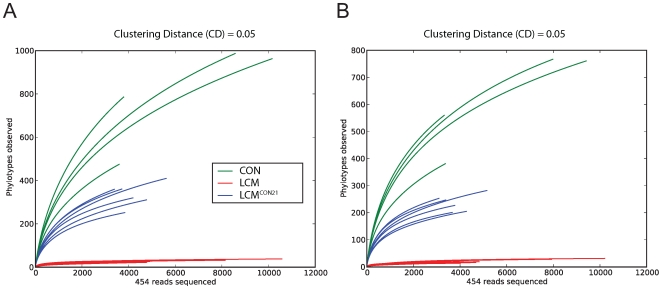
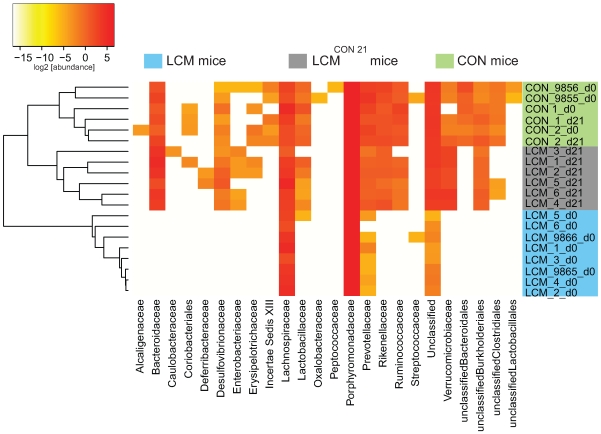
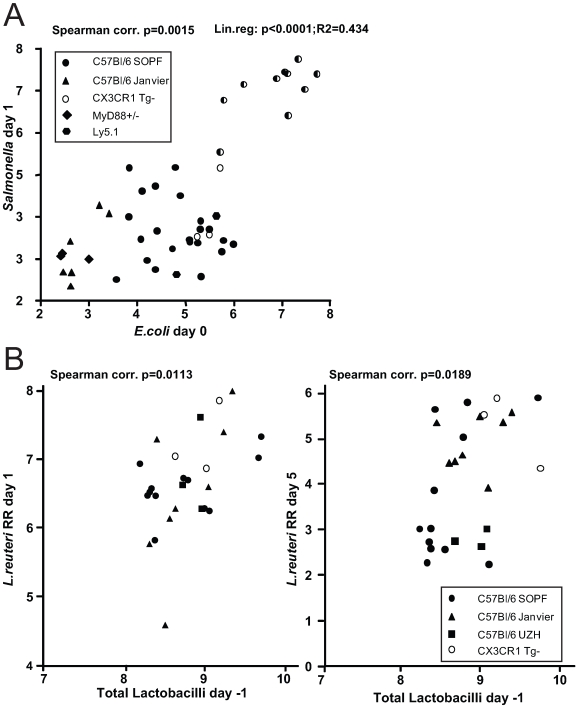
The intestinal ecosystem is formed by a complex, yet highly characteristic microbial community. The parameters defining whether this community permits invasion of a new bacterial species are unclear. In particular, inhibition of enteropathogen infection by the gut microbiota ( = colonization resistance) is poorly understood. To analyze the mechanisms of microbiota-mediated protection from Salmonella enterica induced enterocolitis, we used a mouse infection model and large scale high-throughput pyrosequencing. In contrast to conventional mice (CON), mice with a gut microbiota of low complexity (LCM) were highly susceptible to S. enterica induced colonization and enterocolitis. Colonization resistance was partially restored in LCM-animals by co-housing with conventional mice for 21 days (LCM(con21)). 16S rRNA sequence analysis comparing LCM, LCM(con21) and CON gut microbiota revealed that gut microbiota complexity increased upon conventionalization and correlated with increased resistance to S. enterica infection. Comparative microbiota analysis of mice with varying degrees of colonization resistance allowed us to identify intestinal ecosystem characteristics associated with susceptibility to S. enterica infection. Moreover, this system enabled us to gain further insights into the general principles of gut ecosystem invasion by non-pathogenic, commensal bacteria. Mice harboring high commensal E. coli densities were more susceptible to S. enterica induced gut inflammation. Similarly, mice with high titers of Lactobacilli were more efficiently colonized by a commensal Lactobacillus reuteri(RR) strain after oral inoculation. Upon examination of 16S rRNA sequence data from 9 CON mice we found that closely related phylotypes generally display significantly correlated abundances (co-occurrence), more so than distantly related phylotypes. Thus, in essence, the presence of closely related species can increase the chance of invasion of newly incoming species into the gut ecosystem. We provide evidence that this principle might be of general validity for invasion of bacteria in preformed gut ecosystems. This might be of relevance for human enteropathogen infections as well as therapeutic use of probiotic commensal bacteria.
肠道生态系统由一个复杂但高度特征化的微生物群落组成。目前尚不清楚决定该群落是否允许新的细菌物种入侵的参数。特别是,肠道微生物群(即定植抵抗力)抑制肠道病原体感染的机制还知之甚少。为了分析微生物群介导的对沙门氏菌引起的肠炎的保护机制,我们使用了一种小鼠感染模型和大规模高通量焦磷酸测序。与常规小鼠(CON)相比,具有低复杂性肠道微生物群(LCM)的小鼠极易被沙门氏菌定植并引起肠炎。在与常规小鼠共笼 21 天(LCM(con21))后,LCM 动物的定植抵抗力部分恢复。将 LCM、LCM(con21)和 CON 肠道微生物群进行 16S rRNA 序列分析,结果表明,常规化后肠道微生物群的复杂性增加,并且与对沙门氏菌感染的抵抗力增加相关。对具有不同定植抵抗力的小鼠的微生物群进行比较分析,使我们能够确定与沙门氏菌感染易感性相关的肠道生态系统特征。此外,该系统使我们能够进一步了解非致病性共生细菌入侵肠道生态系统的一般原则。携带高共生大肠杆菌密度的小鼠更容易发生沙门氏菌引起的肠道炎症。同样,在口服接种共生乳酸杆菌 RR 菌株后,高滴度乳酸杆菌的小鼠更容易被定植。在检查 9 只 CON 小鼠的 16S rRNA 序列数据后,我们发现,密切相关的分类群通常显示出显著相关的丰度(共现),而远缘的分类群则不然。因此,从本质上讲,密切相关的物种的存在可以增加新进入的物种入侵肠道生态系统的机会。我们提供的证据表明,这一原则可能对预先形成的肠道生态系统中细菌的入侵具有普遍的有效性。这可能与人类肠道病原体感染以及益生菌共生细菌的治疗应用有关。