Academy of Mathematics and Systems Science, Chinese Academy of Sciences, Beijing 100190, PR China.
BMC Bioinformatics. 2010 Jan 13;11:26. doi: 10.1186/1471-2105-11-26.
The accumulation of high-throughput data greatly promotes computational investigation of gene function in the context of complex biological systems. However, a biological function is not simply controlled by an individual gene since genes function in a cooperative manner to achieve biological processes. In the study of human diseases, rather than to discover disease related genes, identifying disease associated pathways and modules becomes an essential problem in the field of systems biology.
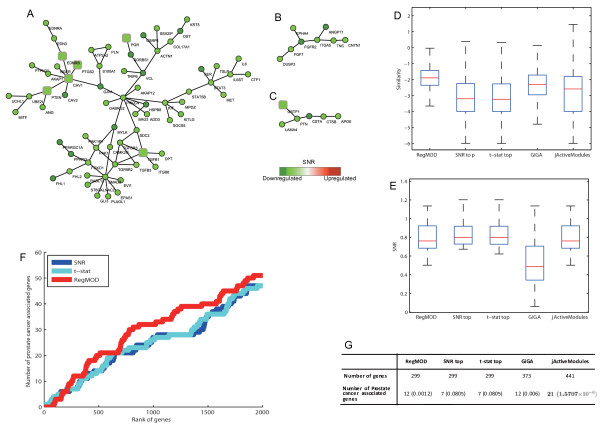
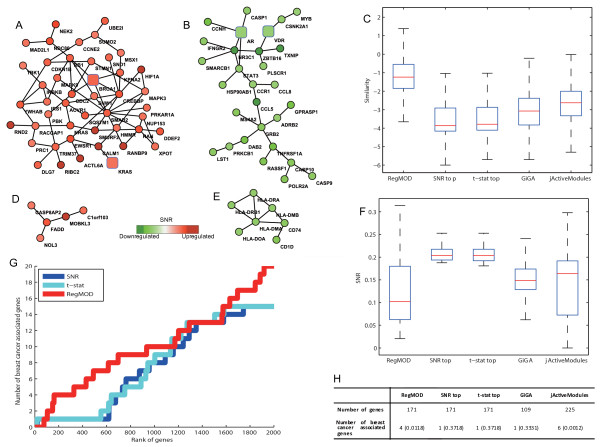
In this paper, we propose a novel method to detect disease related gene modules or dysfunctional pathways based on global characteristics of interactome coupled with gene expression data. Specifically, we exploit interacting relationships between genes to define a gene's active score function based on the kernel trick, which can represent nonlinear effects of gene cooperativity. Then, modules or pathways are inferred based on the active scores evaluated by the support vector regression in a global and integrative manner. The efficiency and robustness of the proposed method are comprehensively validated by using both simulated and real data with the comparison to existing methods.
By applying the proposed method to two cancer related problems, i.e. breast cancer and prostate cancer, we successfully identified active modules or dysfunctional pathways related to these two types of cancers with literature confirmed evidences. We show that this network-based method is highly efficient and can be applied to a large-scale problem especially for human disease related modules or pathway extraction. Moreover, this method can also be used for prioritizing genes associated with a specific phenotype or disease.
高通量数据的积累极大地促进了在复杂生物系统背景下对基因功能的计算研究。然而,由于基因以协作的方式发挥作用来实现生物过程,因此单个基因并不能简单地控制生物功能。在人类疾病的研究中,识别疾病相关的途径和模块,而不是发现与疾病相关的基因,已成为系统生物学领域的一个基本问题。
在本文中,我们提出了一种新的方法,基于互作组的全局特征和基因表达数据来检测疾病相关的基因模块或功能失调的途径。具体来说,我们利用基因之间的相互关系,基于核技巧定义了一个基因的活性评分函数,该函数可以表示基因协同作用的非线性效应。然后,基于支持向量回归在全局和综合的方式评估活性评分来推断模块或途径。通过使用模拟数据和真实数据,并与现有方法进行比较,全面验证了所提出方法的效率和稳健性。
通过将所提出的方法应用于两个癌症相关问题,即乳腺癌和前列腺癌,我们成功地识别了与这两种癌症相关的活性模块或功能失调的途径,这些途径都有文献证实的证据。我们表明,这种基于网络的方法非常高效,可应用于大规模问题,特别是用于提取与人类疾病相关的模块或途径。此外,该方法还可用于对与特定表型或疾病相关的基因进行优先级排序。