Department of Bioinformatics and Genomics, University of North Carolina at Charlotte, Charlotte, North Carolina, United States of America.
PLoS One. 2010 Jan 20;5(1):e8797. doi: 10.1371/journal.pone.0008797.
Transcription factor binding site (TFBS) motifs can be accurately represented by position frequency matrices (PFM) or other equivalent forms. We often need to compare TFBS motifs using their PFMs in order to search for similar motifs in a motif database, or cluster motifs according to their binding preference. The majority of current methods for motif comparison involve a similarity metric for column-to-column comparison and a method to find the optimal position alignment between the two compared motifs. In some applications, alignment-free methods might be preferred; however, few such methods with high accuracy have been described.
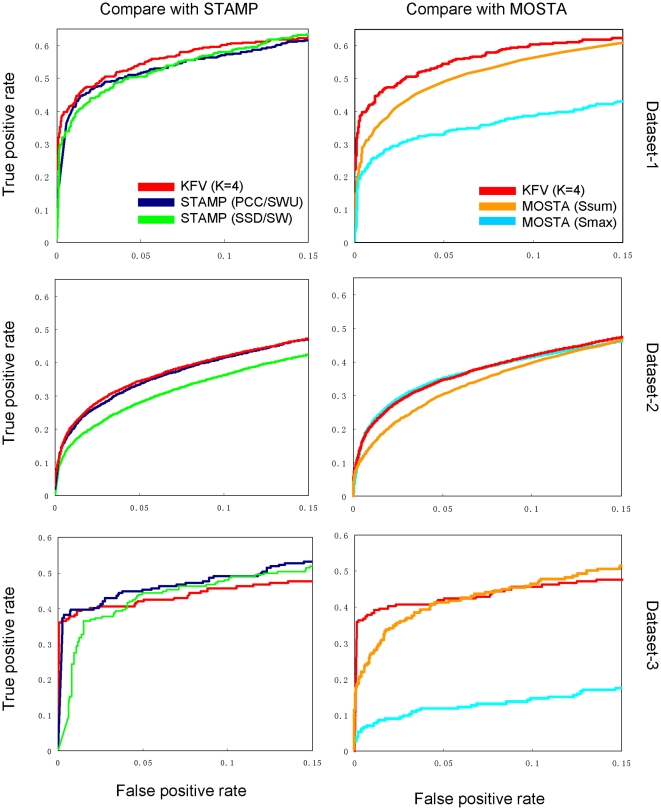
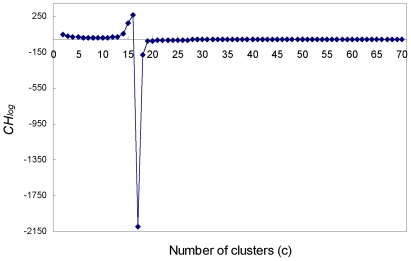
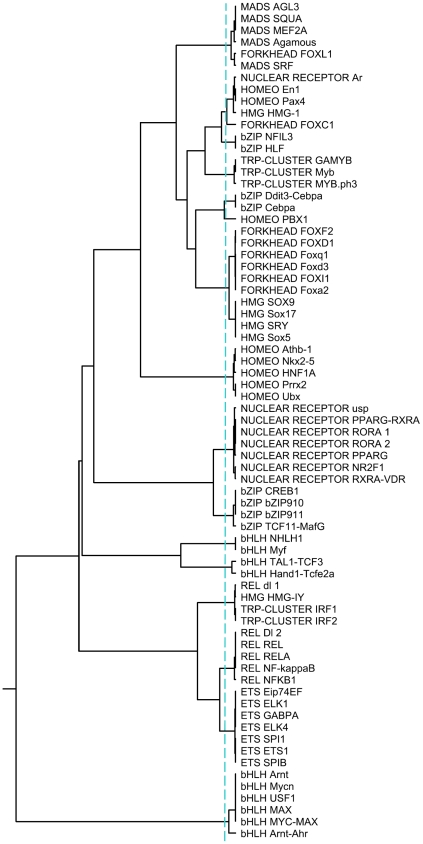
METHODOLOGY/PRINCIPAL FINDINGS: Here we describe a novel alignment-free method for quantifying the similarity of motifs using their PFMs by converting PFMs into k-mer vectors. The motifs could then be compared by measuring the similarity among their corresponding k-mer vectors.
CONCLUSIONS/SIGNIFICANCE: We demonstrate that our method in general achieves similar performance or outperforms the existing methods for clustering motifs according to their binding preference and identifying similar motifs of transcription factors of the same family.
转录因子结合位点(TFBS)基序可以通过位置频率矩阵(PFM)或其他等效形式准确地表示。为了在 motif 数据库中搜索相似的基序,或者根据结合偏好对基序进行聚类,我们经常需要使用它们的 PFM 来比较 TFBS 基序。目前大多数用于 motif 比较的方法都涉及列与列之间比较的相似性度量和在两个比较的基序之间找到最佳位置对齐的方法。在某些应用中,可能更喜欢无对齐方法;但是,已经描述的具有高精度的此类方法很少。
方法/主要发现:在这里,我们描述了一种使用其 PFM 将 PFM 转换为 k-mer 向量来量化基序相似性的新型无对齐方法。然后可以通过测量它们对应的 k-mer 向量之间的相似性来比较基序。
结论/意义:我们证明,我们的方法通常可以根据结合偏好对基序进行聚类,并识别相同家族的转录因子的相似基序,从而实现与现有方法类似的性能,或者优于现有方法。