David R, Cheriton School of Computer Science, University of Waterloo, Waterloo, Ontario, Canada.
BMC Bioinformatics. 2010 Jan 18;11 Suppl 1(Suppl 1):S11. doi: 10.1186/1471-2105-11-S1-S11.
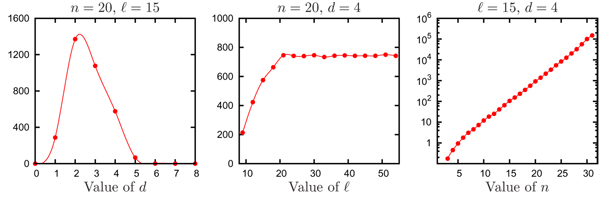
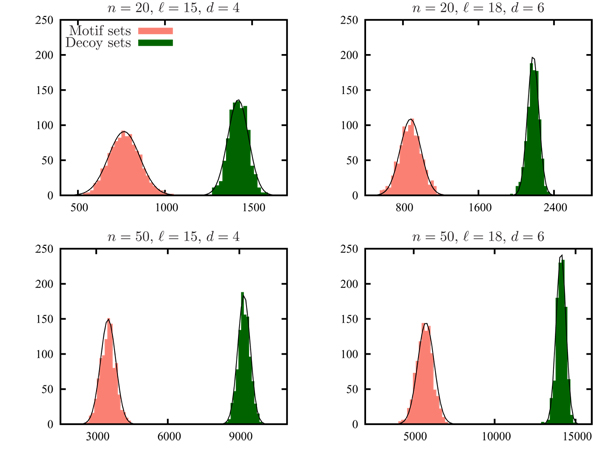
Improving the accuracy and efficiency of motif recognition is an important computational challenge that has application to detecting transcription factor binding sites in genomic data. Closely related to motif recognition is the CONSENSUS STRING decision problem that asks, given a parameter d and a set of l-length strings S = {s1, ..., sn}, whether there exists a consensus string that has Hamming distance at most d from any string in S. A set of strings S is pairwise bounded if the Hamming distance between any pair of strings in S is at most 2d. It is trivial to determine whether a set is pairwise bounded, and a set cannot have a consensus string unless it is pairwise bounded. We use CONSENSUS STRING to determine whether or not a pairwise bounded set has a consensus. Unfortunately, CONSENSUS STRING is NP-complete. The lack of an efficient method to solve the CONSENSUS STRING problem has caused it to become a computational bottleneck in MCL-WMR, a motif recognition program capable of solving difficult motif recognition problem instances.
We focus on the development of a method for solving CONSENSUS STRING quickly with a small probability of error. We apply this heuristic to develop a new motif recognition program, sMCL-WMR, which has impressive accuracy and efficiency. We demonstrate the performance of sMCL-WMR in detecting weak motifs in large data sets and in real genomic data sets, and compare the performance to other leading motif recognition programs. In our preliminary discussion of our CONSENSUS STRING algorithm we give insight into the issue of sampling pairwise bounded sets, and discuss its relevance to motif recognition.
Our novel heuristic gives birth to a state of the art program, sMCL-WMR, that is capable of detecting weak motifs in data sets with a large number of strings. sMCL-WMR is orders of magnitude faster than its predecessor MCL-WMR and is capable of solving previously unsolved synthetic motif recognition problems. Lastly, sMCL-WMR shows impressive accuracy in detecting transcription factor binding sites in the genomic data and used in the assessment of Tompa et al.
提高基序识别的准确性和效率是一个重要的计算挑战,它在检测基因组数据中的转录因子结合位点方面有应用。与基序识别密切相关的是共识字符串决策问题,该问题询问,给定参数 d 和一组 l 长度的字符串 S={s1,…,sn},是否存在一个共识字符串,使得其与 S 中的任何字符串的汉明距离都不超过 d。一组字符串 S 是成对有界的,如果 S 中任意两个字符串之间的汉明距离都不超过 2d。确定一组字符串是否是成对有界的是微不足道的,并且除非集合是成对有界的,否则它不可能有一个共识字符串。我们使用 CONSENSUS STRING 来确定成对有界的集合是否有共识字符串。不幸的是,CONSENSUS STRING 是 NP 完全的。缺乏一种有效的方法来解决 CONSENSUS STRING 问题,导致它成为 MCL-WMR 中的一个计算瓶颈,MCL-WMR 是一个能够解决困难基序识别问题实例的基序识别程序。
我们专注于开发一种快速解决 CONSENSUS STRING 问题的方法,同时具有较小的错误概率。我们将此启发式方法应用于开发一个新的基序识别程序 sMCL-WMR,该程序具有令人印象深刻的准确性和效率。我们在大型数据集和真实基因组数据集中检测弱基序的性能,并将性能与其他领先的基序识别程序进行比较。在我们对 CONSENSUS STRING 算法的初步讨论中,我们深入了解了对成对有界集合进行抽样的问题,并讨论了它与基序识别的相关性。
我们的新启发式方法孕育了一个最先进的程序 sMCL-WMR,它能够在具有大量字符串的数据集检测弱基序。sMCL-WMR 比其前身 MCL-WMR 快几个数量级,并且能够解决以前无法解决的合成基序识别问题。最后,sMCL-WMR 在检测基因组数据中的转录因子结合位点和用于评估 Tompa 等人的实验数据方面表现出令人印象深刻的准确性。