Centre for Cellular and Molecular Biology, Hyderabad, India.
BMC Bioinformatics. 2010 Jan 18;11 Suppl 1(Suppl 1):S26. doi: 10.1186/1471-2105-11-S1-S26.
Human Immunodeficiency Virus type 1 (HIV-1), the causative agent of Acquired Immune Deficiency Syndrome (AIDS), exhibits very high genetic diversity with different variants or subtypes prevalent in different parts of the world. Proper classification of the HIV-1 subtypes, displaying differential infectivity, plays a major role in monitoring the epidemic and is also a critical component for effective treatment strategy. The existing methods to classify HIV-1 sequence subtypes, based on phylogenetic analysis focusing only on specific genes/regions, have shown inconsistencies as they lack the capability to analyse whole genome variations. Several isolates are left unclassified due to unresolved sub-typing. It is apparent that classification of subtypes based on complete genome sequences, rather than sub-genomic regions, is a more robust and comprehensive approach to address genome-wide heterogeneity. However, no simple methodology exists that directly computes HIV-1 subtype from the complete genome sequence.
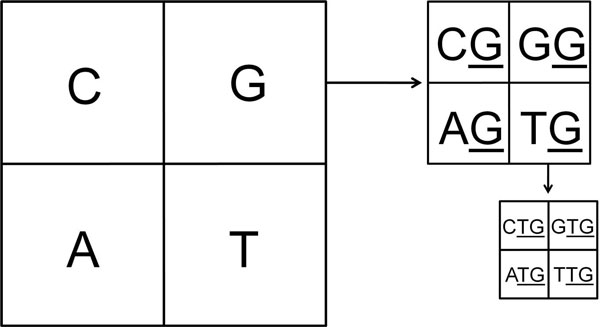
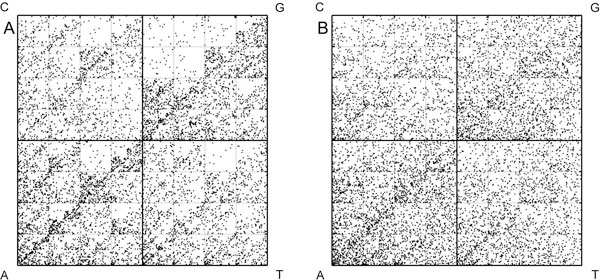
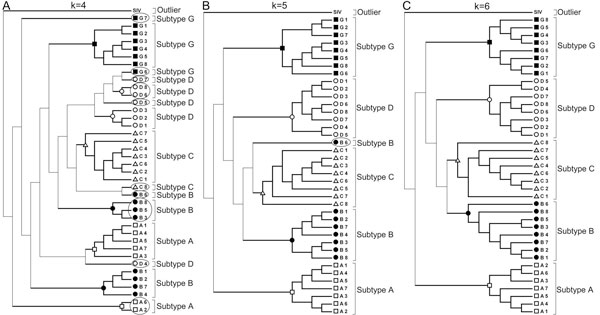
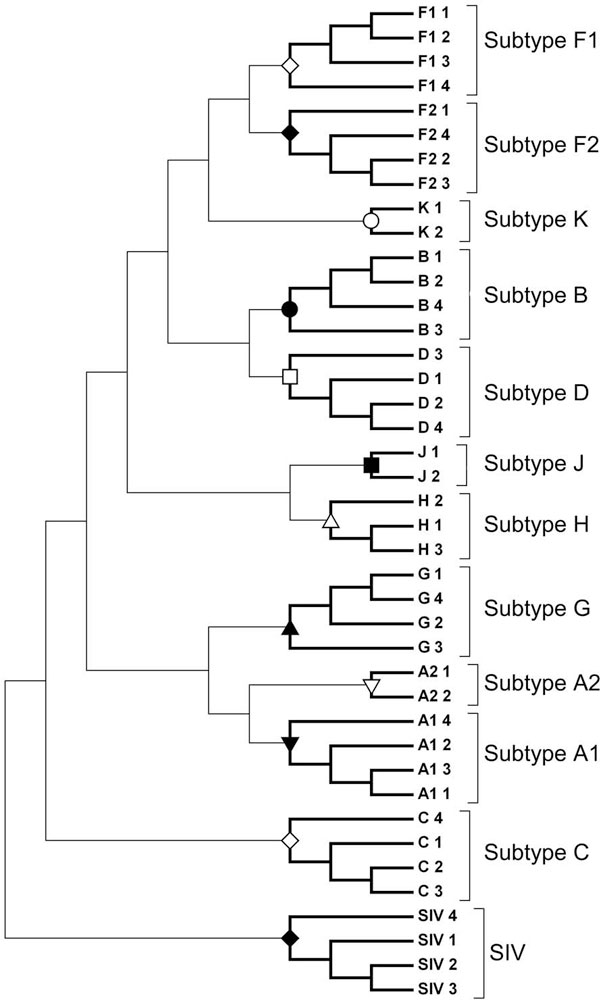
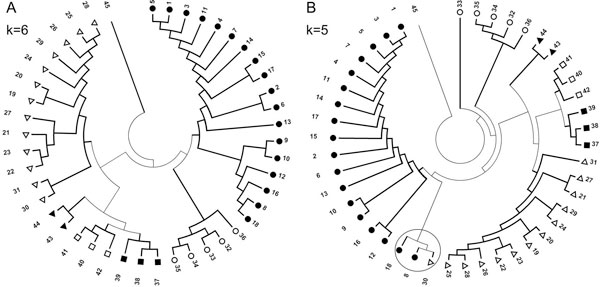
We use Chaos Game Representation (CGR) as an approach to identify the distinctive genomic signature associated with the DNA sequence organisation in different HIV-1 subtypes. We first analysed the effect of nucleotide word lengths (k = 2 to 8) on whole genomes of the HIV-1 M group sequences, and found the optimum word length of k = 6, that could classify HIV-1 subtypes based on a Test sequence set. Using the optimised word length, we then showed accurate classification of the HIV-1 subtypes from both the Reference Set sequences and from all available sequences in the database. Finally, we applied the approach to cluster the five unclassified HIV-1 sequences from Africa and Europe, and predict their possible subtypes.
We propose a genomic signature-based approach, using CGR with suitable word length optimisation, which can be applied to classify intra-species variations, and apply it to the complex problem of HIV-1 subtype classification. We demonstrate that CGR is a simple and computationally less intensive method that not only accurately segregates the HIV-1 subtype and sub-subtypes, but also aid in the classification of the unclassified sequences. We hope that it will be useful in subtype annotation of the newly sequenced HIV-1 genomes.
人类免疫缺陷病毒 1 型(HIV-1)是获得性免疫缺陷综合征(AIDS)的病原体,具有非常高的遗传多样性,不同的变体或亚型在世界不同地区流行。对 HIV-1 亚型进行正确分类,显示出不同的感染性,在监测流行情况方面起着重要作用,也是制定有效治疗策略的关键组成部分。目前基于仅关注特定基因/区域的系统发育分析来对 HIV-1 序列亚型进行分类的方法,由于缺乏分析全基因组变异的能力,因此存在不一致性。由于无法进行亚型细分,有几种分离株未被分类。显然,基于完整基因组序列而不是亚基因组区域对亚型进行分类,是一种更稳健和全面的方法,可以解决全基因组异质性问题。然而,目前尚无直接从完整基因组序列计算 HIV-1 亚型的简单方法。
我们使用混沌游戏表示(CGR)作为一种方法来识别与不同 HIV-1 亚型中 DNA 序列组织相关的独特基因组特征。我们首先分析了核苷酸字长(k = 2 到 8)对 HIV-1 M 组序列的全基因组的影响,发现最优字长 k = 6,可以基于测试序列集对 HIV-1 亚型进行分类。使用优化的字长,我们然后展示了对参考集序列和数据库中所有可用序列的 HIV-1 亚型的准确分类。最后,我们将该方法应用于聚类来自非洲和欧洲的 5 个未分类的 HIV-1 序列,并预测它们可能的亚型。
我们提出了一种基于基因组特征的方法,使用 CGR 并进行适当的字长优化,可以应用于分类种内变异,并将其应用于 HIV-1 亚型分类这一复杂问题。我们证明 CGR 是一种简单且计算强度较低的方法,不仅可以准确地分离 HIV-1 亚型和亚亚型,还可以辅助未分类序列的分类。我们希望它对新测序的 HIV-1 基因组的亚型注释有用。