Laboratory for Computational Biology and Bioinformatics, Swiss Federal Institute of Technology (EPFL), EPFL-IIS-LCBB, INJ 230, Station 14, CH-1015 Lausanne, Switzerland.
BMC Bioinformatics. 2010 Jan 18;11 Suppl 1(Suppl 1):S54. doi: 10.1186/1471-2105-11-S1-S54.
The rapidly increasing availability of whole-genome sequences has enabled the study of whole-genome evolution. Evolutionary mechanisms based on genome rearrangements have attracted much attention and given rise to many models; somewhat independently, the mechanisms of gene duplication and loss have seen much work. However, the two are not independent and thus require a unified treatment, which remains missing to date. Moreover, existing rearrangement models do not fit the dichotomy between most prokaryotic genomes (one circular chromosome) and most eukaryotic genomes (multiple linear chromosomes).
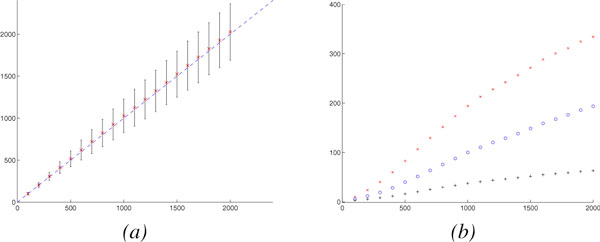
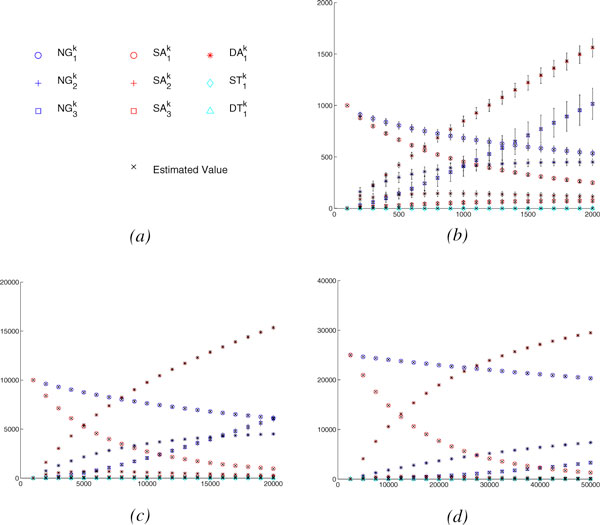
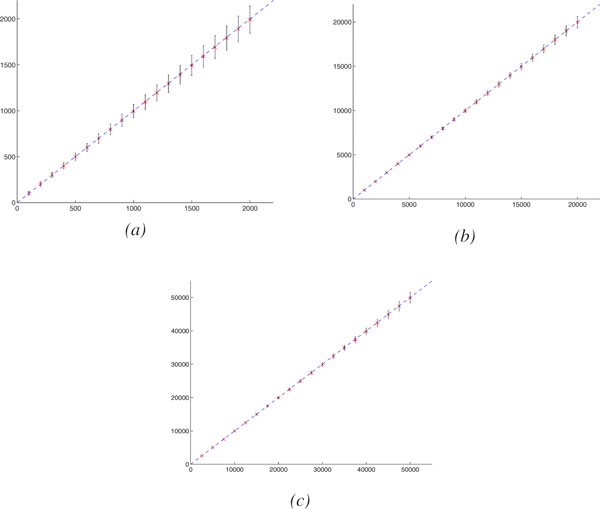
To handle rearrangements, gene duplications and losses, we propose a new evolutionary model and the corresponding method for estimating true evolutionary distance. Our model, inspired from the DCJ model, is simple and the first to respect the prokaryotic/eukaryotic structural dichotomy. Experimental results on a wide variety of genome structures demonstrate the very high accuracy and robustness of our distance estimator.
We give the first robust, statistically based, estimate of genomic pairwise distances based on rearrangements, duplications and losses, under a model that respects the structural dichotomy between prokaryotic and eukaryotic genomes. Accurate and robust estimates in true evolutionary distances should translate into much better phylogenetic reconstructions as well as more accurate genomic alignments, while our new model of genome rearrangements provides another refinement in simplicity and verisimilitude.
全基因组序列的快速普及使得对全基因组进化的研究成为可能。基于基因组重排的进化机制引起了广泛关注,并产生了许多模型;在某种程度上,基因复制和丢失的机制也得到了广泛的研究。然而,这两者并非独立的,因此需要统一处理,但目前仍缺乏这种处理方法。此外,现有的重排模型并不符合大多数原核生物基因组(一个环状染色体)和大多数真核生物基因组(多个线性染色体)之间的二分法。
为了处理重排、基因复制和丢失,我们提出了一种新的进化模型和相应的方法来估计真实的进化距离。我们的模型受 DCJ 模型启发,简单且是第一个尊重原核生物/真核生物结构二分法的模型。在广泛的基因组结构上的实验结果表明,我们的距离估计器具有非常高的准确性和鲁棒性。
我们在尊重原核生物和真核生物基因组结构二分法的模型下,给出了第一个基于重排、复制和丢失的稳健、基于统计学的基因组对距离估计。在真实进化距离上进行准确和稳健的估计应该会转化为更好的系统发育重建以及更准确的基因组比对,而我们的新基因组重排模型在简单性和真实性方面又提供了另一种改进。