Alexeev Nikita, Alekseyev Max A
Computational Biology Institute at the George Washington University, Ashburn, 20147, VA, USA.
BMC Genomics. 2017 May 24;18(Suppl 4):356. doi: 10.1186/s12864-017-3733-3.
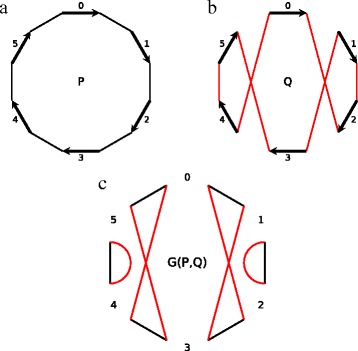
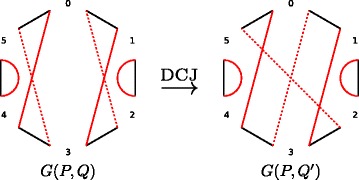
The ability to estimate the evolutionary distance between extant genomes plays a crucial role in many phylogenomic studies. Often such estimation is based on the parsimony assumption, implying that the distance between two genomes can be estimated as the rearrangement distance equal the minimal number of genome rearrangements required to transform one genome into the other. However, in reality the parsimony assumption may not always hold, emphasizing the need for estimation that does not rely on the rearrangement distance. The distance that accounts for the actual (rather than minimal) number of rearrangements between two genomes is often referred to as the true evolutionary distance. While there exists a method for the true evolutionary distance estimation, it however assumes that genomes can be broken by rearrangements equally likely at any position in the course of evolution. This assumption, known as the random breakage model, has recently been refuted in favor of the more rigorous fragile breakage model postulating that only certain "fragile" genomic regions are prone to rearrangements.
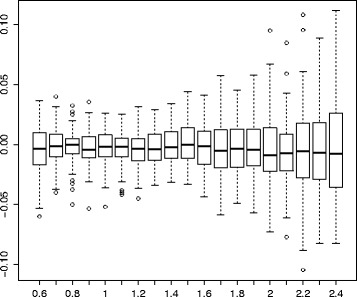
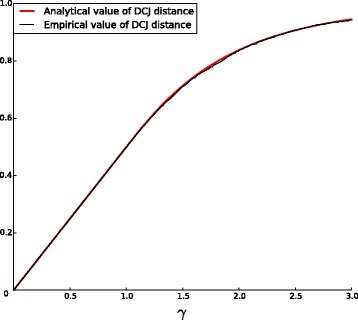
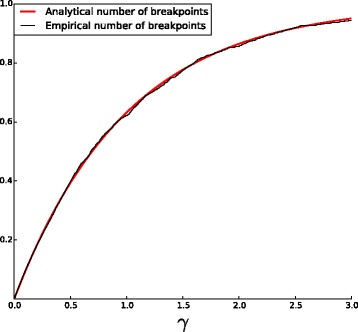
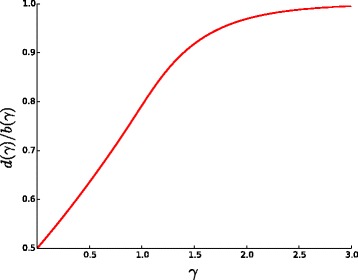
We propose a new method for estimating the true evolutionary distance between two genomes under the fragile breakage model. We evaluate the proposed method on simulated genomes, which show its high accuracy. We further apply the proposed method for estimation of evolutionary distances within a set of five yeast genomes and a set of two fish genomes.
The true evolutionary distances between the five yeast genomes estimated with the proposed method reveals that some pairs of yeast genomes violate the parsimony assumption. The proposed method further demonstrates that the rearrangement distance between the two fish genomes underestimates their evolutionary distance by about 20%. These results demonstrate how drastically the two distances can differ and justify the use of true evolutionary distance in phylogenomic studies.
在许多系统发育基因组学研究中,估计现存基因组之间的进化距离的能力起着至关重要的作用。通常,这种估计基于简约假设,这意味着两个基因组之间的距离可以估计为等于将一个基因组转化为另一个基因组所需的最小基因组重排数的重排距离。然而,在现实中,简约假设可能并不总是成立,这就强调了需要不依赖于重排距离的估计方法。考虑两个基因组之间实际(而非最小)重排数的距离通常被称为真实进化距离。虽然存在一种用于估计真实进化距离的方法,但它假设基因组在进化过程中的任何位置被重排打断的可能性相同。这个假设,即随机断裂模型,最近已被反驳,转而支持更严格的脆弱断裂模型,该模型假定只有某些“脆弱”的基因组区域容易发生重排。
我们提出了一种在脆弱断裂模型下估计两个基因组之间真实进化距离的新方法。我们在模拟基因组上评估了所提出的方法,结果显示其具有很高的准确性。我们进一步将所提出的方法应用于一组五个酵母基因组和一组两个鱼类基因组的进化距离估计。
用所提出的方法估计的五个酵母基因组之间的真实进化距离表明,一些酵母基因组对违反了简约假设。所提出的方法进一步证明,两个鱼类基因组之间的重排距离低估了它们的进化距离约20%。这些结果表明这两种距离可能有多大的差异,并证明在系统发育基因组学研究中使用真实进化距离是合理的。