Stanford Genome Technology Center, 955 California Avenue, Palo Alto, California, 94305, USA.
BMC Bioinformatics. 2010 Jan 18;11 Suppl 1(Suppl 1):S8. doi: 10.1186/1471-2105-11-S1-S8.
The large amount of high-throughput genomic data has facilitated the discovery of the regulatory relationships between transcription factors and their target genes. While early methods for discovery of transcriptional regulation relationships from microarray data often focused on the high-throughput experimental data alone, more recent approaches have explored the integration of external knowledge bases of gene interactions.
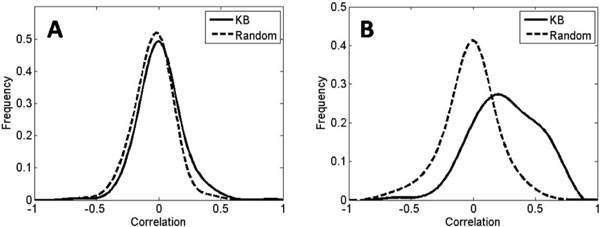
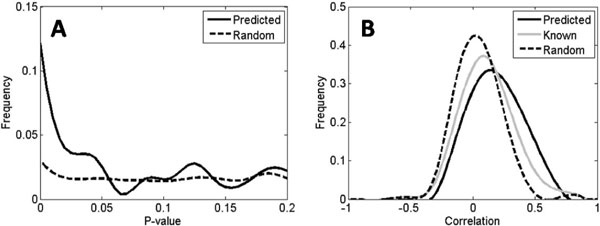
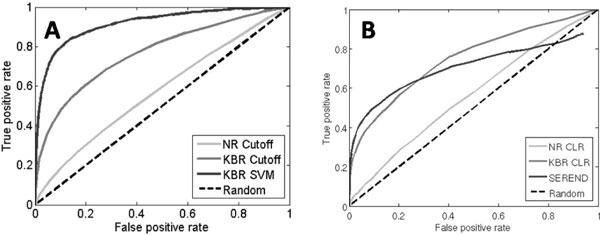
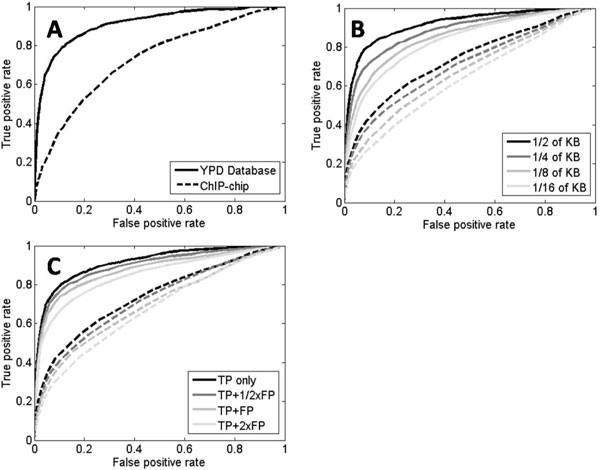
In this work, we develop an algorithm that provides improved performance in the prediction of transcriptional regulatory relationships by supplementing the analysis of microarray data with a new method of integrating information from an existing knowledge base. Using a well-known dataset of yeast microarrays and the Yeast Proteome Database, a comprehensive collection of known information of yeast genes, we show that knowledge-based predictions demonstrate better sensitivity and specificity in inferring new transcriptional interactions than predictions from microarray data alone. We also show that comprehensive, direct and high-quality knowledge bases provide better prediction performance. Comparison of our results with ChIP-chip data and growth fitness data suggests that our predicted genome-wide regulatory pairs in yeast are reasonable candidates for follow-up biological verification.
High quality, comprehensive, and direct knowledge bases, when combined with appropriate bioinformatic algorithms, can significantly improve the discovery of gene regulatory relationships from high throughput gene expression data.
大量高通量基因组数据促进了转录因子与其靶基因之间调控关系的发现。虽然早期从微阵列数据中发现转录调控关系的方法通常仅侧重于高通量实验数据,但最近的方法已经探索了整合基因相互作用的外部知识库。
在这项工作中,我们开发了一种算法,通过用一种从现有知识库中集成信息的新方法来补充微阵列数据分析,从而在预测转录调控关系方面提供了更好的性能。使用酵母微阵列的著名数据集和酵母蛋白质组数据库,这是酵母基因的已知信息的综合收集,我们表明,基于知识的预测在推断新的转录相互作用方面比单独使用微阵列数据的预测具有更好的敏感性和特异性。我们还表明,全面、直接和高质量的知识库提供了更好的预测性能。与 ChIP-chip 数据和生长适应性数据的比较表明,我们在酵母中预测的全基因组调控对是后续生物学验证的合理候选者。
高质量、全面和直接的知识库,结合适当的生物信息学算法,可以显著提高从高通量基因表达数据中发现基因调控关系的能力。