Yang Bei, Xu Yaohui, Maxwell Andrew, Koh Wonryull, Gong Ping, Zhang Chaoyang
School of Information & Engineering, Zhengzhou University, Zhengzhou, 450000, China.
Center of Precision Medicine, Zhengzhou University, Zhengzhou, 450000, China.
BMC Syst Biol. 2018 Dec 14;12(Suppl 7):115. doi: 10.1186/s12918-018-0635-1.
Reconstruction of gene regulatory networks (GRNs), also known as reverse engineering of GRNs, aims to infer the potential regulation relationships between genes. With the development of biotechnology, such as gene chip microarray and RNA-sequencing, the high-throughput data generated provide us with more opportunities to infer the gene-gene interaction relationships using gene expression data and hence understand the underlying mechanism of biological processes. Gene regulatory networks are known to exhibit a multiplicity of interaction mechanisms which include functional and non-functional, and linear and non-linear relationships. Meanwhile, the regulatory interactions between genes and gene products are not spontaneous since various processes involved in producing fully functional and measurable concentrations of transcriptional factors/proteins lead to a delay in gene regulation. Many different approaches for reconstructing GRNs have been proposed, but the existing GRN inference approaches such as probabilistic Boolean networks and dynamic Bayesian networks have various limitations and relatively low accuracy. Inferring GRNs from time series microarray data or RNA-sequencing data remains a very challenging inverse problem due to its nonlinearity, high dimensionality, sparse and noisy data, and significant computational cost, which motivates us to develop more effective inference methods.
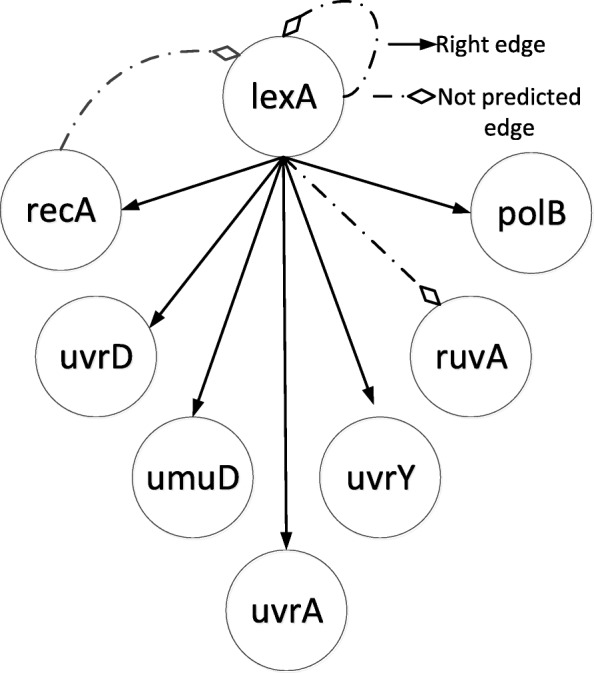
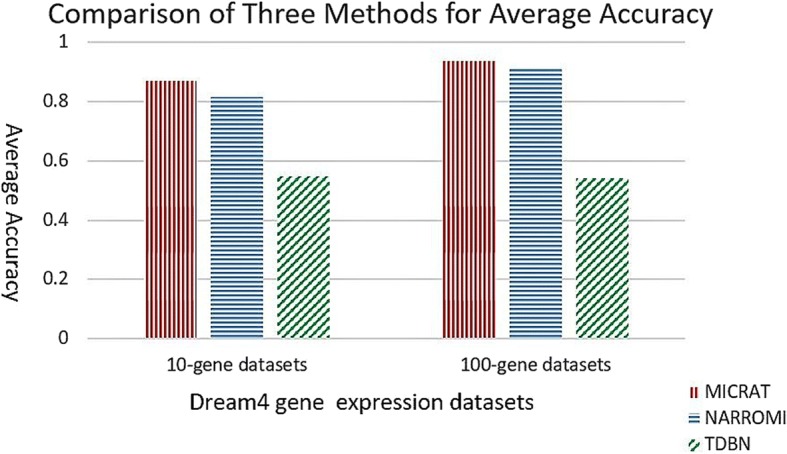
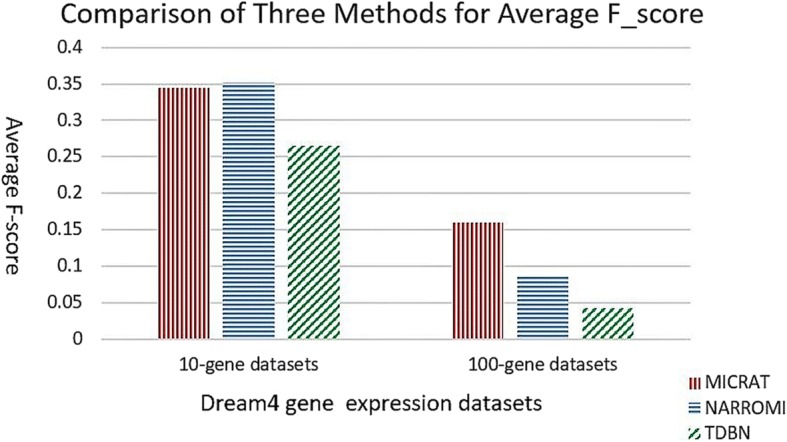
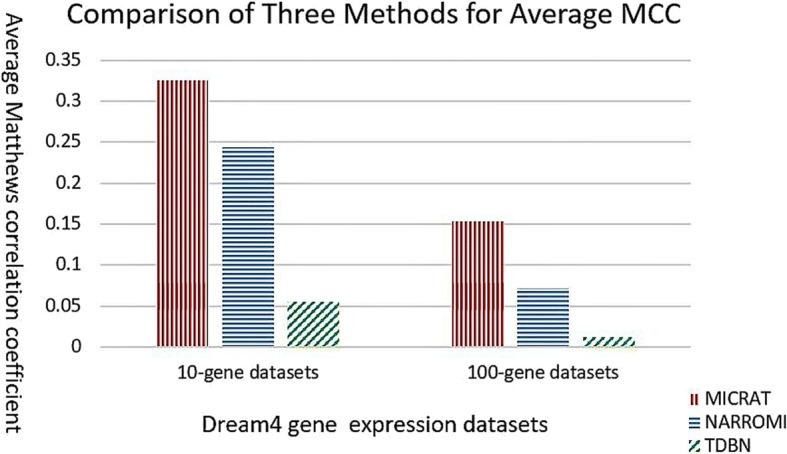
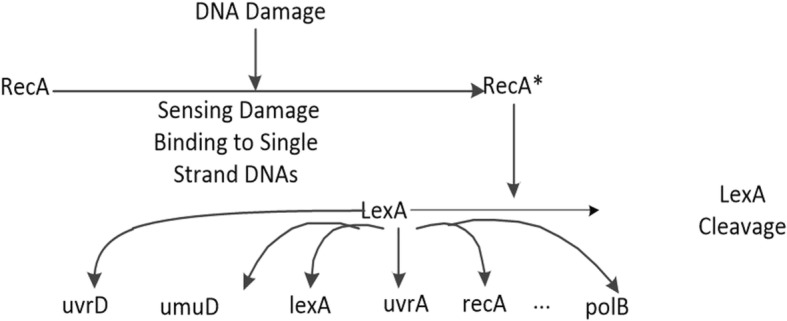
We developed a novel algorithm, MICRAT (Maximal Information coefficient with Conditional Relative Average entropy and Time-series mutual information), for inferring GRNs from time series gene expression data. Maximal information coefficient (MIC) is an effective measure of dependence for two-variable relationships. It captures a wide range of associations, both functional and non-functional, and thus has good performance on measuring the dependence between two genes. Our approach mainly includes two procedures. Firstly, it employs maximal information coefficient for constructing an undirected graph to represent the underlying relationships between genes. Secondly, it directs the edges in the undirected graph for inferring regulators and their targets. In this procedure, the conditional relative average entropies of each pair of nodes (or genes) are employed to indicate the directions of edges. Since the time delay might exist in the expression of regulators and target genes, time series mutual information is combined to cooperatively direct the edges for inferring the potential regulators and their targets. We evaluated the performance of MICRAT by applying it to synthetic datasets as well as real gene expression data and compare with other GRN inference methods. We inferred five 10-gene and five 100-gene networks from the DREAM4 challenge that were generated using the gene expression simulator GeneNetWeaver (GNW). MICRAT was also used to reconstruct GRNs on real gene expression data including part of the DNA-damaged response pathway (SOS DNA repair network) and experimental dataset in E. Coli. The results showed that MICRAT significantly improved the inference accuracy, compared to other inference methods, such as TDBN, etc. CONCLUSION: In this work, a novel algorithm, MICRAT, for inferring GRNs from time series gene expression data was proposed by taking into account dependence and time delay of expressions of a regulator and its target genes. This approach employed maximal information coefficients for reconstructing an undirected graph to represent the underlying relationships between genes. The edges were directed by combining conditional relative average entropy with time course mutual information of pairs of genes. The proposed algorithm was evaluated on the benchmark GRNs provided by the DREAM4 challenge and part of the real SOS DNA repair network in E. Coli. The experimental study showed that our approach was comparable to other methods on 10-gene datasets and outperformed other methods on 100-gene datasets in GRN inference from time series datasets.
基因调控网络(GRN)的重建,也称为GRN的逆向工程,旨在推断基因之间潜在的调控关系。随着生物技术的发展,如基因芯片微阵列和RNA测序,所产生的高通量数据为我们提供了更多机会,利用基因表达数据推断基因-基因相互作用关系,从而理解生物过程的潜在机制。已知基因调控网络表现出多种相互作用机制,包括功能性和非功能性、线性和非线性关系。同时,基因与基因产物之间的调控相互作用并非自发产生,因为产生完全功能性和可测量浓度的转录因子/蛋白质所涉及的各种过程会导致基因调控出现延迟。已经提出了许多不同的重建GRN的方法,但现有的GRN推断方法,如概率布尔网络和动态贝叶斯网络,存在各种局限性且准确性相对较低。从时间序列微阵列数据或RNA测序数据推断GRN仍然是一个极具挑战性的逆问题,因为它具有非线性、高维度、数据稀疏且有噪声以及计算成本高昂等特点,这促使我们开发更有效的推断方法。
我们开发了一种名为MICRAT(具有条件相对平均熵和时间序列互信息的最大信息系数)的新算法,用于从时间序列基因表达数据推断GRN。最大信息系数(MIC)是衡量双变量关系依赖性的有效指标。它能捕捉广泛的关联,包括功能性和非功能性关联,因此在测量两个基因之间的依赖性方面表现良好。我们的方法主要包括两个步骤。首先,它采用最大信息系数构建一个无向图来表示基因之间的潜在关系。其次,它对无向图中的边进行定向,以推断调控因子及其靶标。在这个步骤中,利用每对节点(或基因)的条件相对平均熵来指示边的方向。由于调控因子和靶标基因的表达可能存在时间延迟,因此结合时间序列互信息来协同定向边,以推断潜在的调控因子及其靶标。我们通过将MICRAT应用于合成数据集以及真实基因表达数据来评估其性能,并与其他GRN推断方法进行比较。我们从使用基因表达模拟器GeneNetWeaver(GNW)生成的DREAM4挑战中推断出五个10基因和五个100基因的网络。MICRAT还用于在真实基因表达数据上重建GRN,包括部分DNA损伤反应途径(SOS DNA修复网络)和大肠杆菌中的实验数据集。结果表明,与其他推断方法(如TDBN等)相比,MICRAT显著提高了推断准确性。
在这项工作中,通过考虑调控因子及其靶标基因表达的依赖性和时间延迟,提出了一种用于从时间序列基因表达数据推断GRN的新算法MICRAT。该方法采用最大信息系数重建无向图来表示基因之间的潜在关系。通过将条件相对平均熵与基因对的时间进程互信息相结合来定向边。在DREAM4挑战提供的基准GRN以及大肠杆菌中部分真实的SOS DNA修复网络上对所提出的算法进行了评估。实验研究表明,在从时间序列数据集推断GRN方面,我们的方法在10基因数据集上与其他方法相当,在100基因数据集上优于其他方法。