Huang He, Ozkirimli Elif, Post Carol Beth
Department of Medicinal Chemistry and Molecular Pharmacology, Markey Center for Structural Biology and Purdue Cancer Center, Purdue University, West Lafayette, IN, 47907, USA.
J Chem Theory Comput. 2009 Apr 9;5(5):1301-1314. doi: 10.1021/ct9000153.
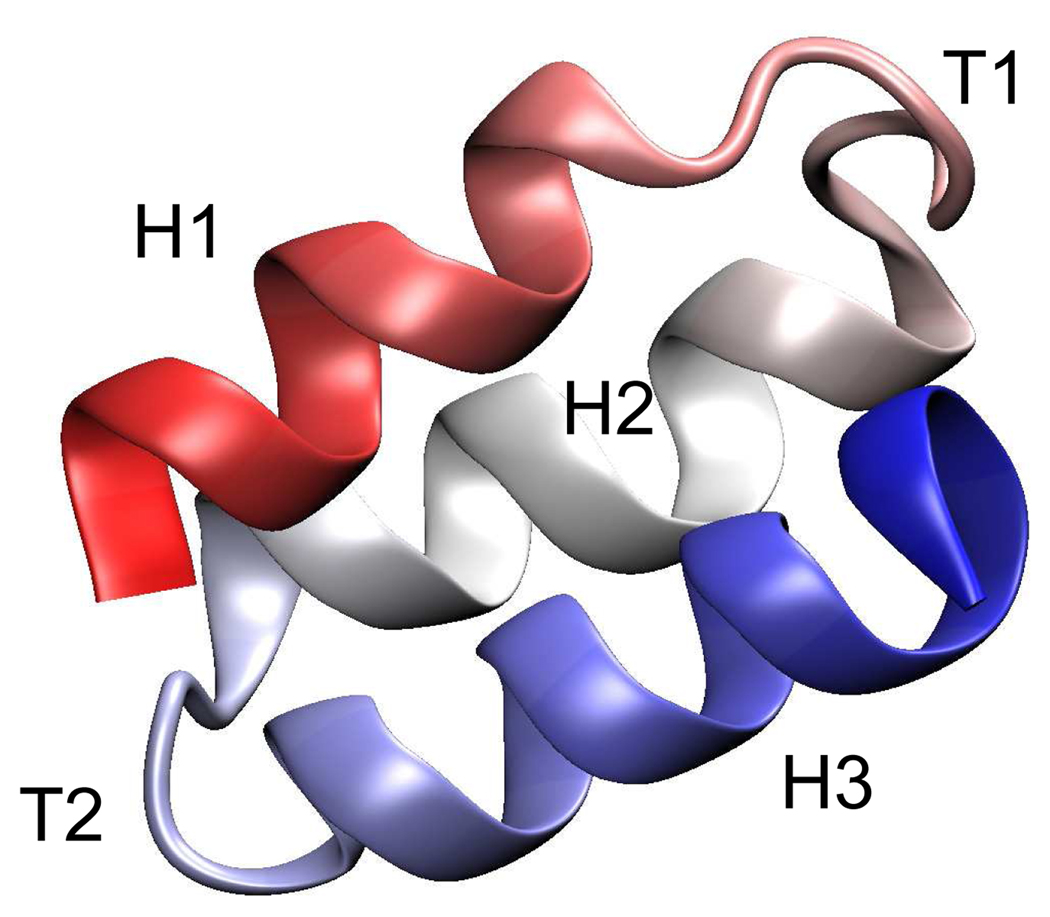
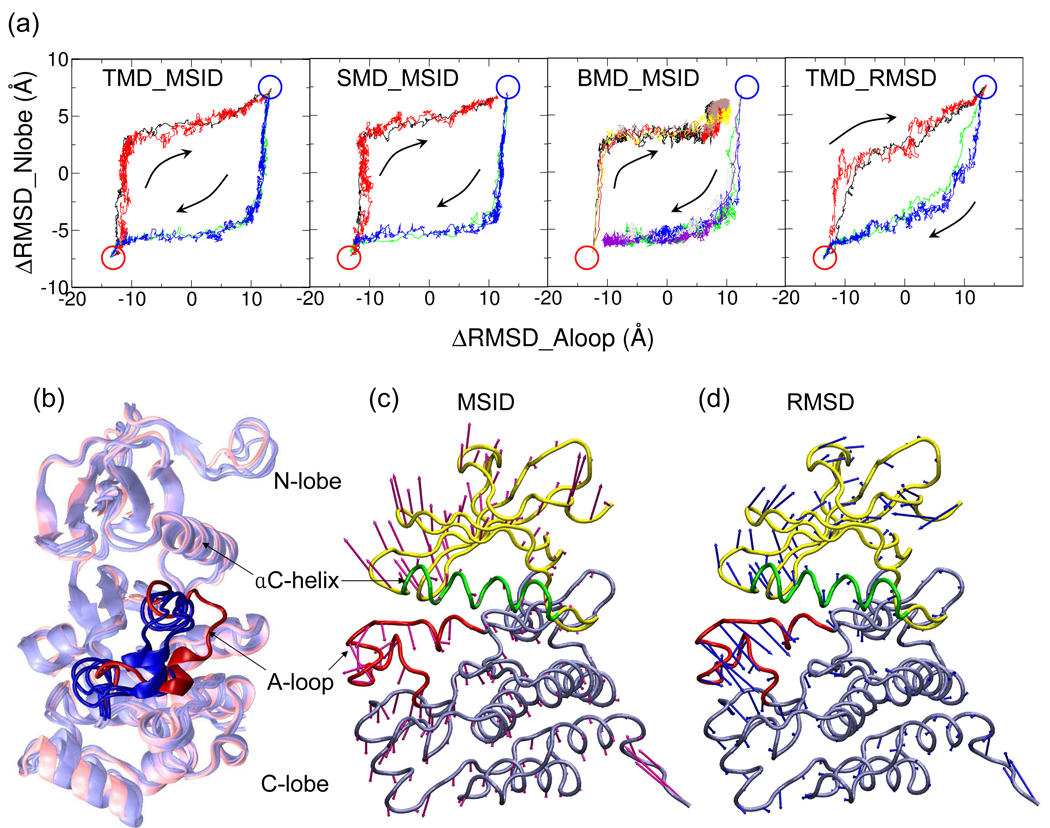
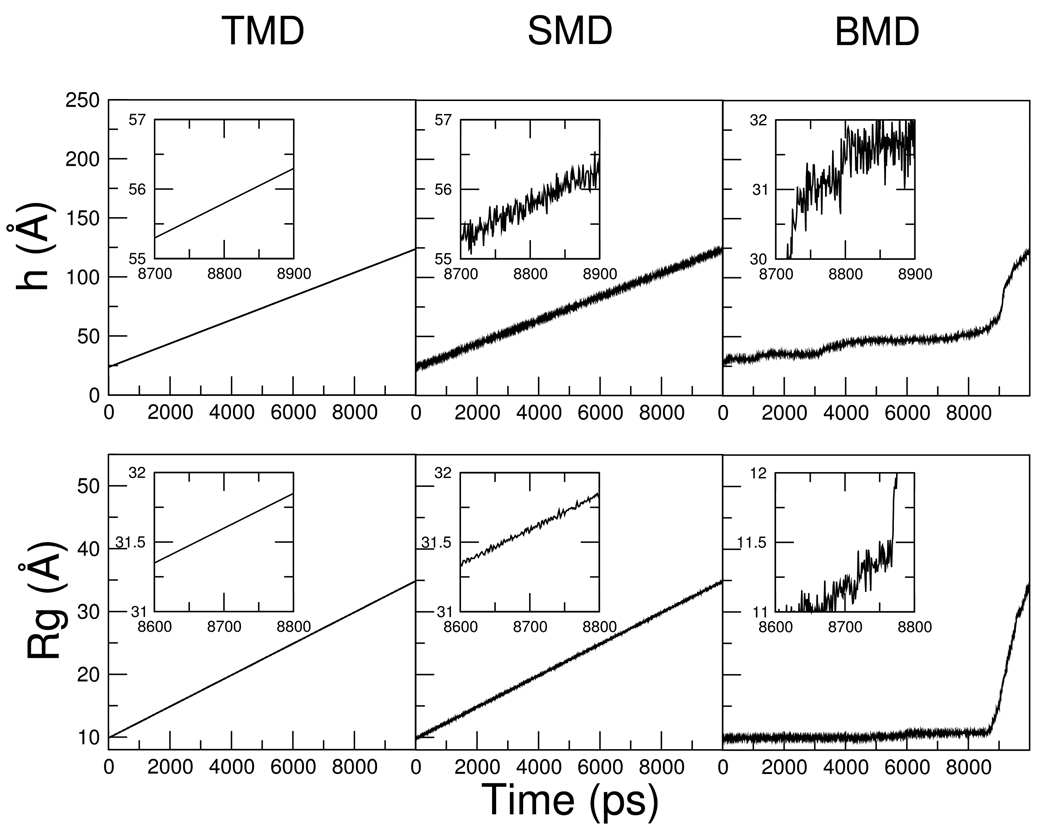
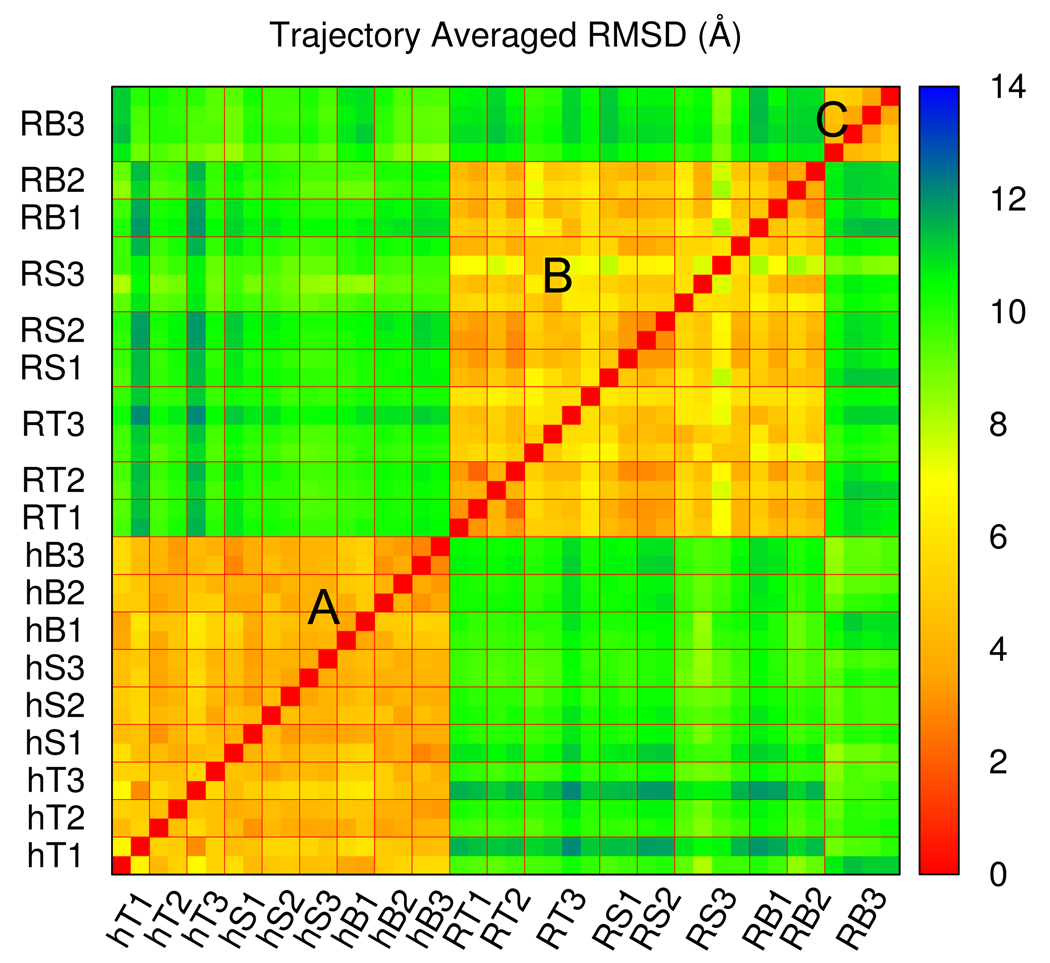
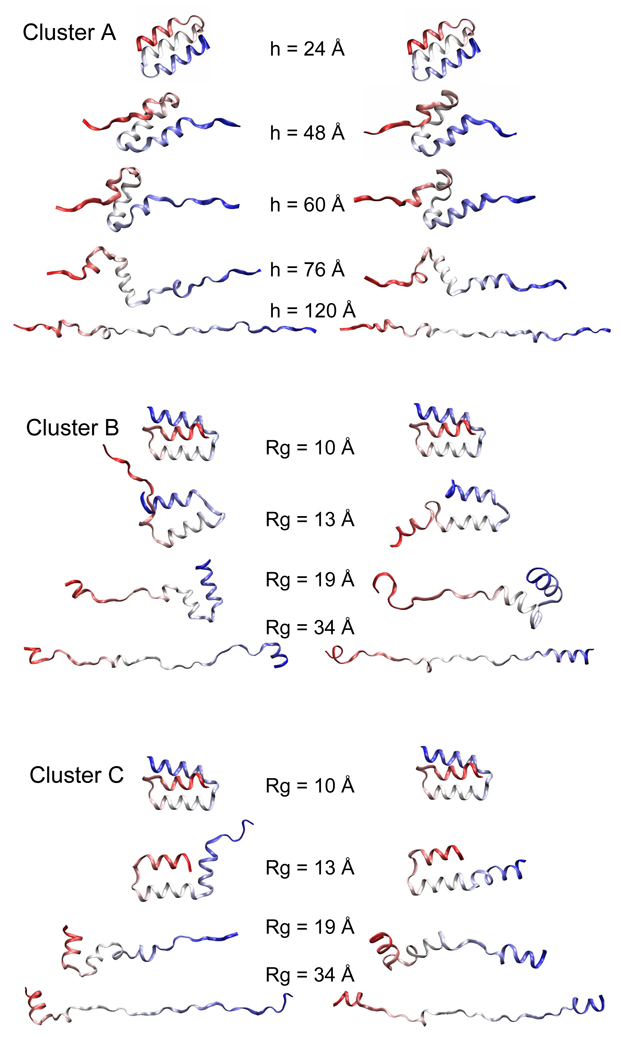
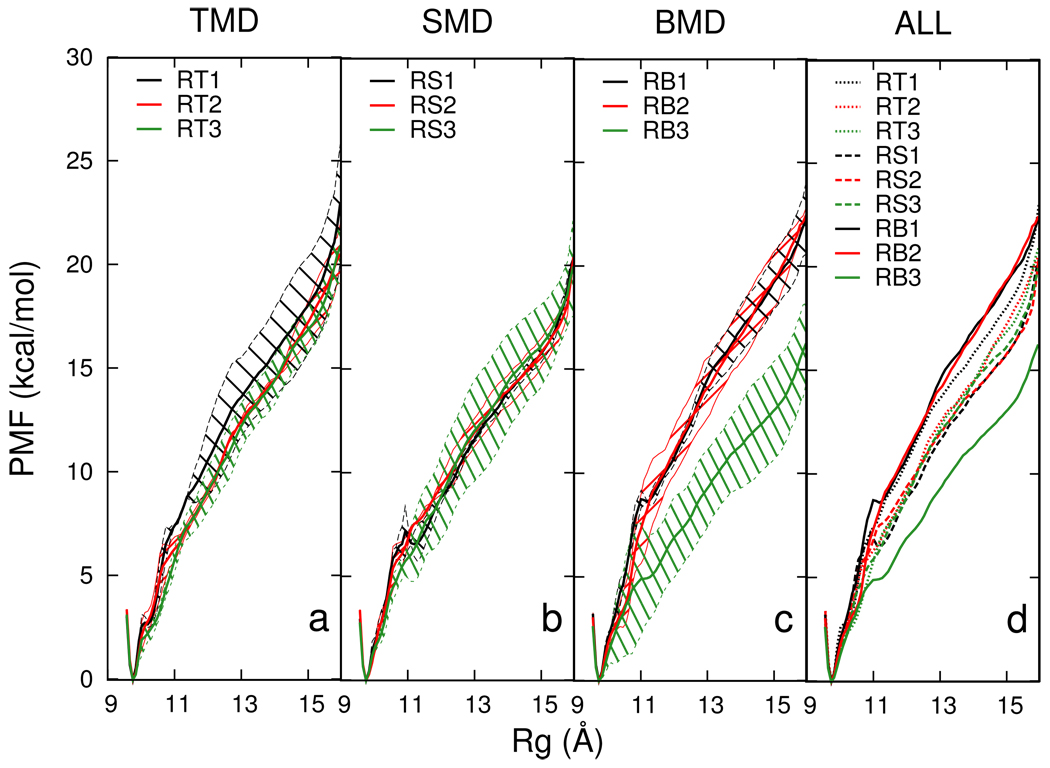
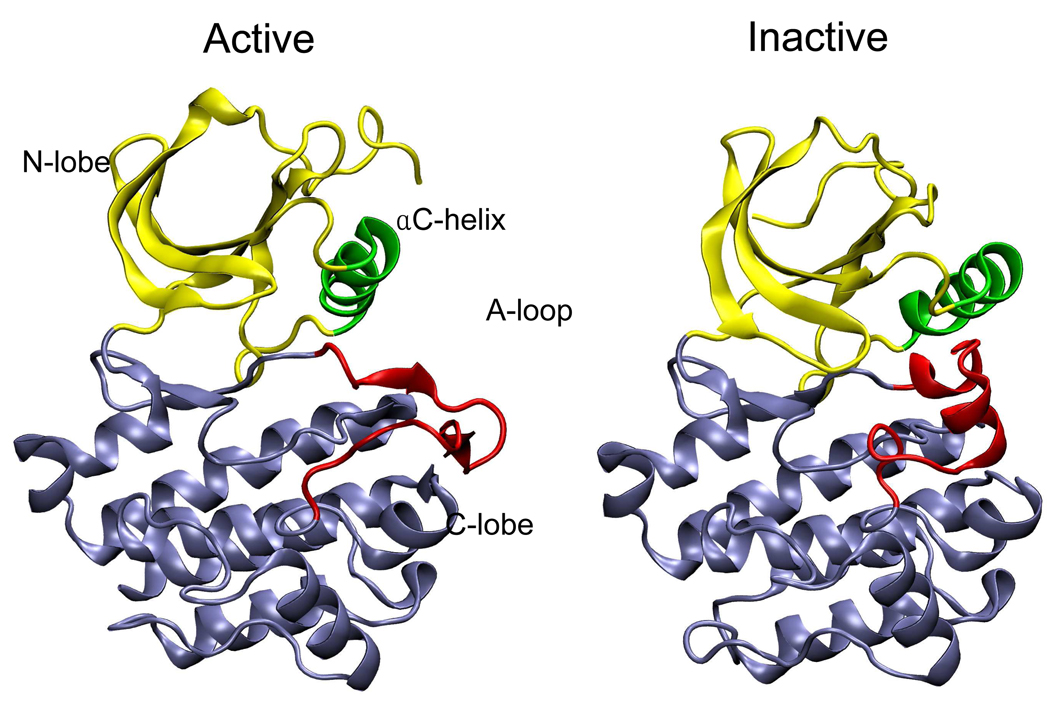
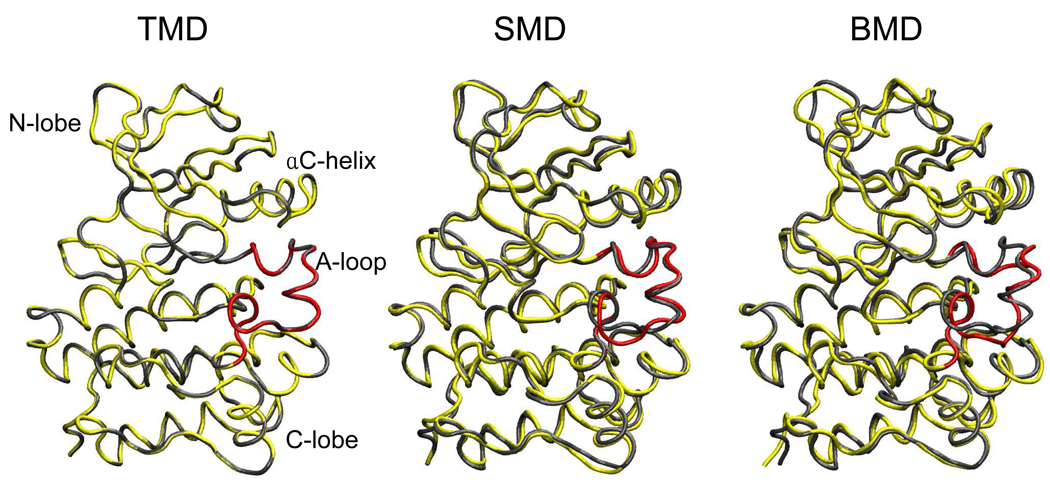
Targeted, steered, and biased molecular dynamics (MD) are widely used methods for studying transition processes of biomolecules. They share the common feature of adding external perturbations along a conformational progress variable to guide the transition in a predefined direction in conformational space, yet differ in how these perturbations are applied. In the present paper, we report a comparison of these three methods on generating transition paths for two different processes: the unfolding of the B domain of protein A and a conformational transition of the catalytic domain of a Src kinase Lyn. Transition pathways were calculated with different simulation parameters including the choice of progress variable and the simulation length or biasing force constant. A comparison of the generated paths based on structural similarity finds that the three perturbation MD methods generate similar transition paths for a given progress variable in most cases. On the other hand, the path depends more strongly on the choice of progress variable used to move the system between the initial and final states. Potentials of mean force (PMF) were calculated starting from unfolding trajectories to estimate the relative probabilities of the paths. A lower PMF was found for the lowest biasing force constant with BMD.
靶向、引导和有偏分子动力学(MD)是研究生物分子过渡过程的广泛使用的方法。它们的共同特点是沿着构象进展变量添加外部扰动,以在构象空间中沿预定义方向引导过渡,但在应用这些扰动的方式上有所不同。在本文中,我们报告了这三种方法在为两个不同过程生成过渡路径方面的比较:蛋白质A的B结构域的去折叠以及Src激酶Lyn的催化结构域的构象转变。使用不同的模拟参数计算过渡路径,包括进展变量的选择以及模拟长度或偏置力常数。基于结构相似性对生成路径的比较发现,在大多数情况下,对于给定的进展变量,三种扰动MD方法生成相似的过渡路径。另一方面,路径更强烈地依赖于用于在初始状态和最终状态之间移动系统的进展变量的选择。从去折叠轨迹开始计算平均力势(PMF),以估计路径的相对概率。发现使用BMD时,最低偏置力常数对应的PMF较低。