Department of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, United States.
J Phys Chem B. 2012 Jul 26;116(29):8584-603. doi: 10.1021/jp212634z. Epub 2012 Mar 28.
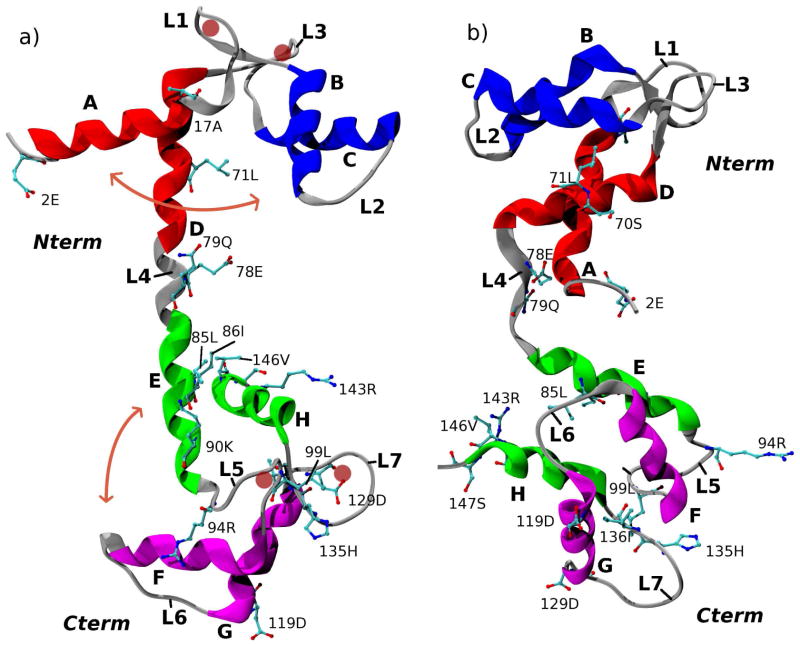
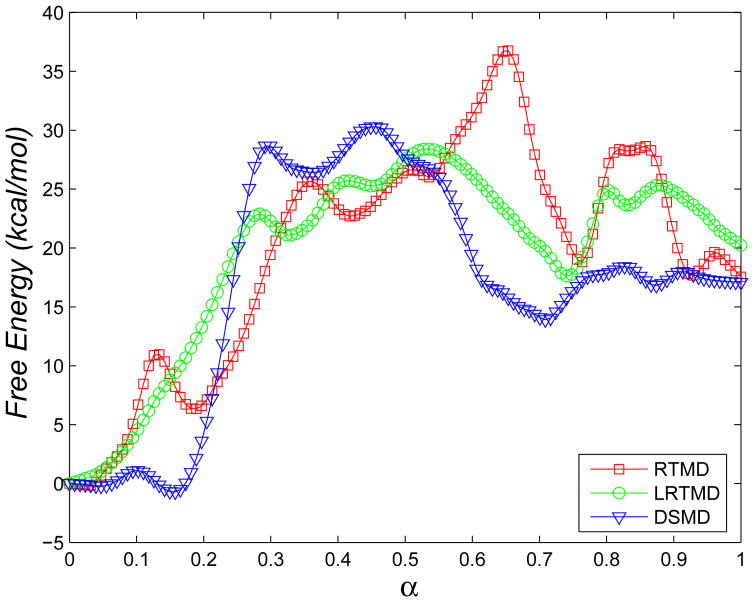
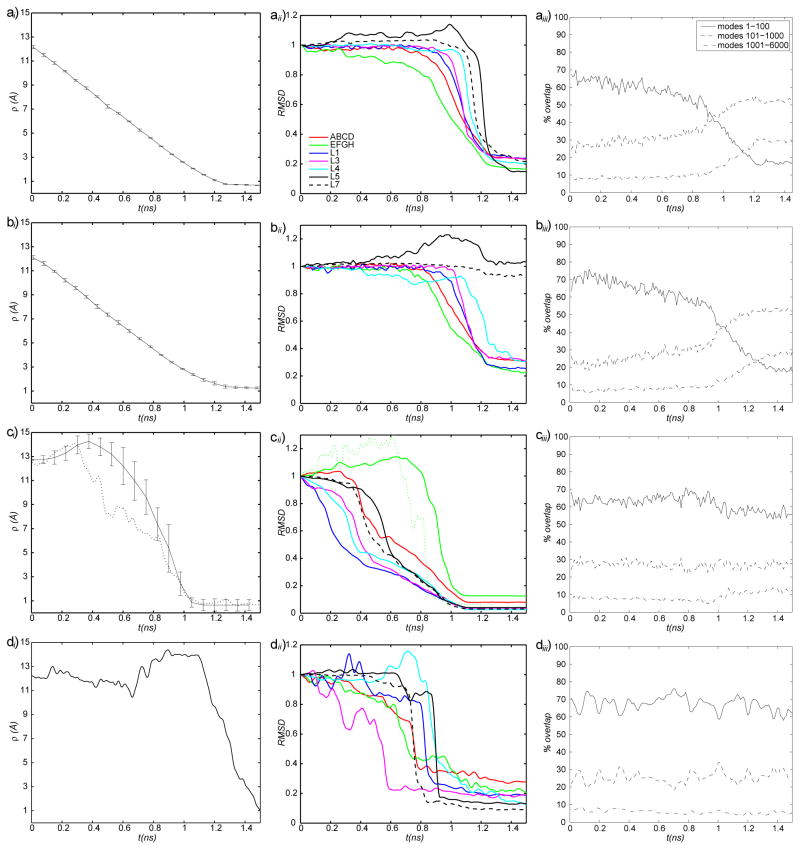
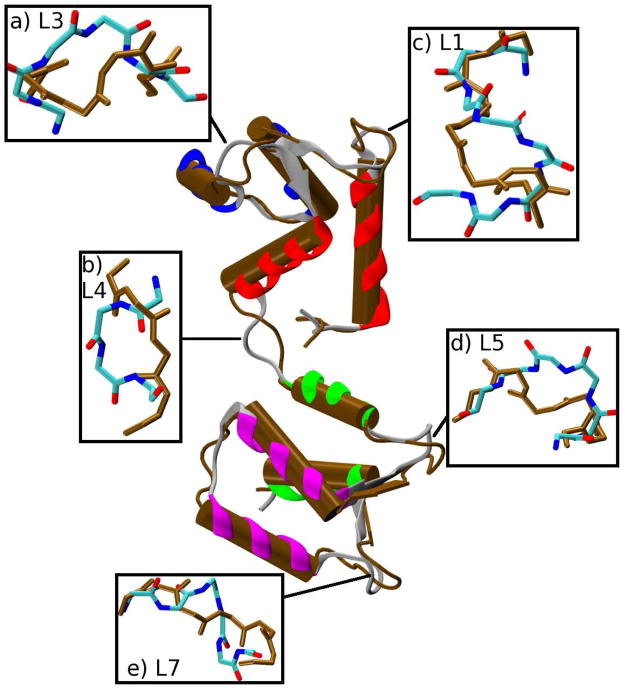
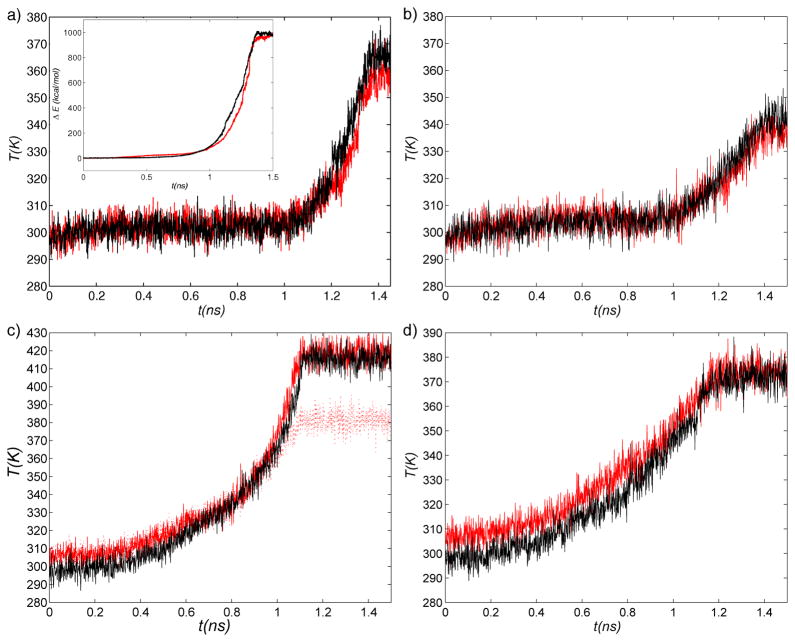
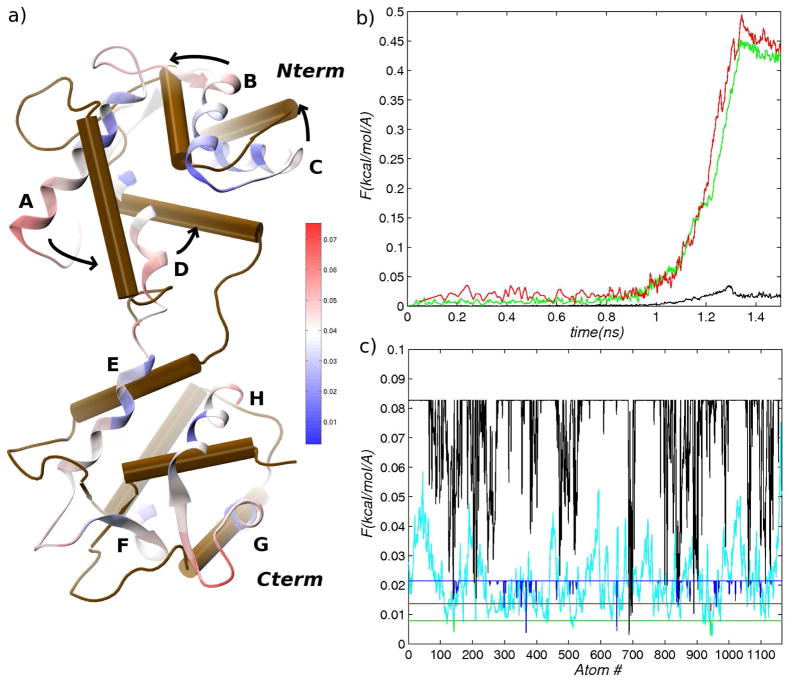
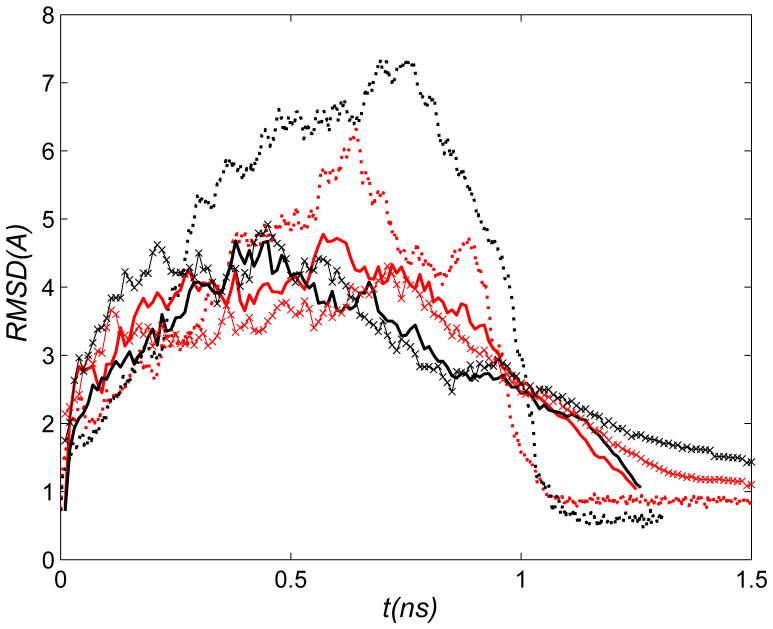
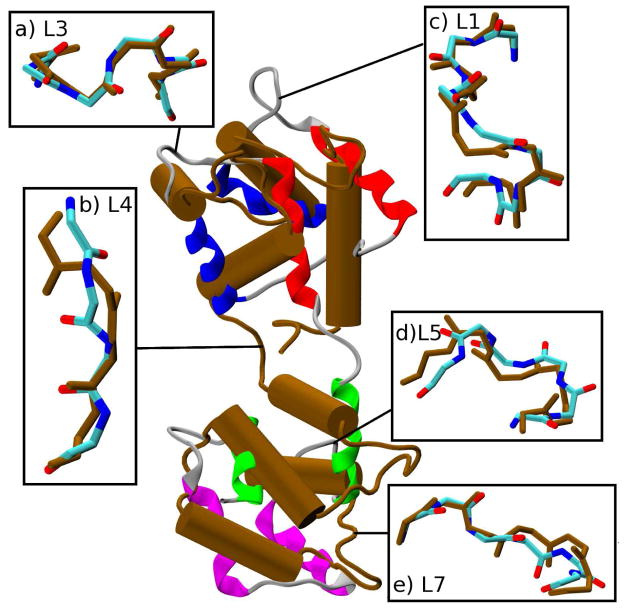
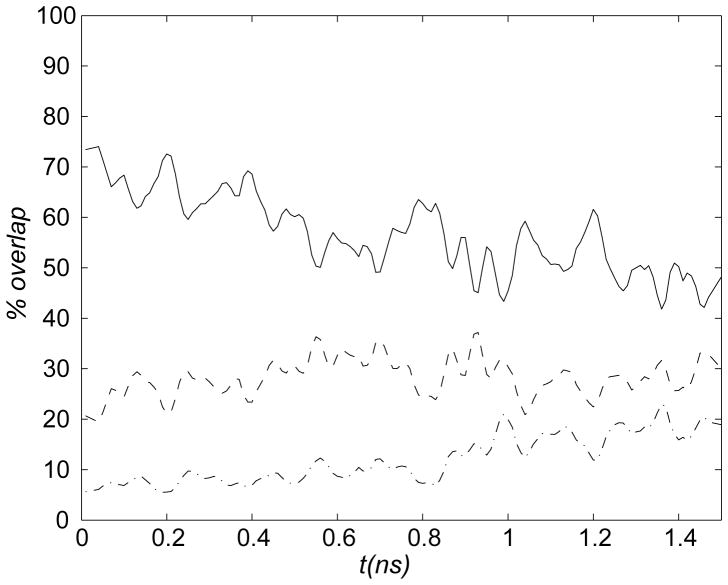
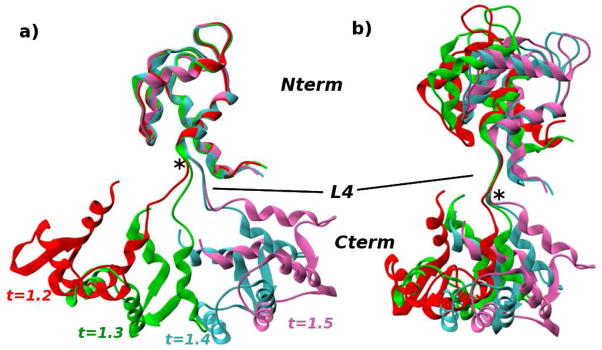
The popular targeted molecular dynamics (TMD) method for generating transition paths in complex biomolecular systems is revisited. In a typical TMD transition path, the large-scale changes occur early and the small-scale changes tend to occur later. As a result, the order of events in the computed paths depends on the direction in which the simulations are performed. To identify the origin of this bias, and to propose a method in which the bias is absent, variants of TMD in the restraint formulation are introduced and applied to the complex open ↔ closed transition in the protein calmodulin. Due to the global best-fit rotation that is typically part of the TMD method, the simulated system is guided implicitly along the lowest-frequency normal modes, until the large spatial scales associated with these modes are near the target conformation. The remaining portion of the transition is described progressively by higher-frequency modes, which correspond to smaller-scale rearrangements. A straightforward modification of TMD that avoids the global best-fit rotation is the locally restrained TMD (LRTMD) method, in which the biasing potential is constructed from a number of TMD potentials, each acting on a small connected portion of the protein sequence. With a uniform distribution of these elements, transition paths that lack the length-scale bias are obtained. Trajectories generated by steered MD in dihedral angle space (DSMD), a method that avoids best-fit rotations altogether, also lack the length-scale bias. To examine the importance of the paths generated by TMD, LRTMD, and DSMD in the actual transition, we use the finite-temperature string method to compute the free energy profile associated with a transition tube around a path generated by each algorithm. The free energy barriers associated with the paths are comparable, suggesting that transitions can occur along each route with similar probabilities. This result indicates that a broad ensemble of paths needs to be calculated to obtain a full description of conformational changes in biomolecules. The breadth of the contributing ensemble suggests that energetic barriers for conformational transitions in proteins are offset by entropic contributions that arise from a large number of possible paths.
重新探讨了用于生成复杂生物分子体系中转变途径的流行靶向分子动力学(TMD)方法。在典型的 TMD 转变途径中,大尺度变化发生得较早,小尺度变化往往发生得较晚。因此,计算途径中的事件顺序取决于模拟进行的方向。为了确定这种偏差的来源,并提出一种不存在偏差的方法,我们引入了约束形式的 TMD 变体,并将其应用于蛋白质钙调蛋白的开放到闭合的复杂转变。由于 TMD 方法通常包含全局最佳拟合旋转,因此模拟系统会隐式地沿着最低频率的正则模态引导,直到与这些模态相关的大空间尺度接近目标构象。转变的其余部分逐步由更高频率的模态描述,这些模态对应于更小尺度的重排。避免全局最佳拟合旋转的 TMD 的直接修改是局部约束 TMD(LRTMD)方法,其中偏置势由多个 TMD 势构建,每个势作用于蛋白质序列的一小部分连接区域。通过这些元素的均匀分布,可以获得没有长度尺度偏差的转变途径。完全避免最佳拟合旋转的二面角空间导向 MD(DSMD)生成的轨迹也没有长度尺度偏差。为了检查 TMD、LRTMD 和 DSMD 生成的途径在实际转变中的重要性,我们使用有限温度字符串方法计算与每个算法生成的途径周围的转变管相关的自由能分布。与途径相关的自由能势垒相当,这表明可以沿每个途径以相似的概率发生转变。该结果表明,需要计算广泛的途径集合以获得生物分子构象变化的完整描述。贡献途径的广泛性表明,蛋白质构象转变的能垒被来自大量可能途径的熵贡献抵消。