Department of Computational Biology and Applied Algorithmics, Max Planck Institute for Informatics, 66123 Saarbruecken, Germany.
BMC Bioinformatics. 2010 Feb 17;11:90. doi: 10.1186/1471-2105-11-90.
Experimental screening of large sets of peptides with respect to their MHC binding capabilities is still very demanding due to the large number of possible peptide sequences and the extensive polymorphism of the MHC proteins. Therefore, there is significant interest in the development of computational methods for predicting the binding capability of peptides to MHC molecules, as a first step towards selecting peptides for actual screening.
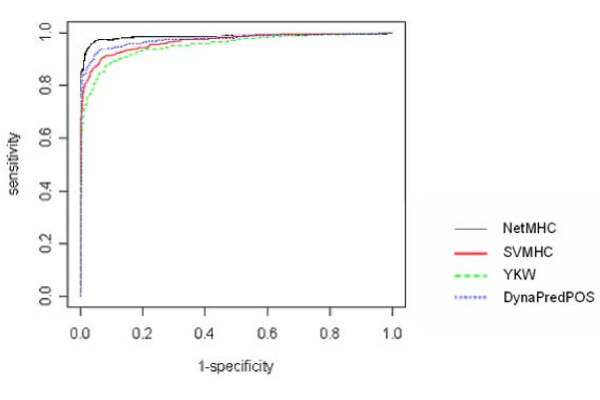
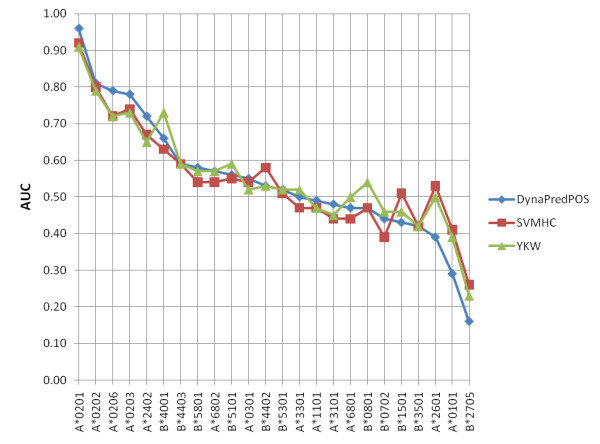
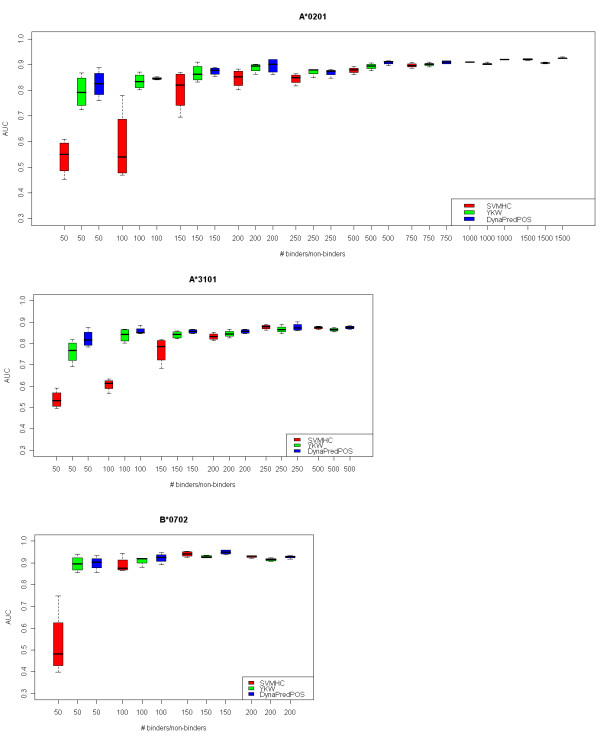
We have examined the performance of four diverse MHC Class I prediction methods on comparatively large HLA-A and HLA-B allele peptide binding datasets extracted from the Immune Epitope Database and Analysis resource (IEDB). The chosen methods span a representative cross-section of available methodology for MHC binding predictions. Until the development of IEDB, such an analysis was not possible, as the available peptide sequence datasets were small and spread out over many separate efforts. We tested three datasets which differ in the IC50 cutoff criteria used to select the binders and non-binders. The best performance was achieved when predictions were performed on the dataset consisting only of strong binders (IC50 less than 10 nM) and clear non-binders (IC50 greater than 10,000 nM). In addition, robustness of the predictions was only achieved for alleles that were represented with a sufficiently large (greater than 200), balanced set of binders and non-binders.
All four methods show good to excellent performance on the comprehensive datasets, with the artificial neural networks based method outperforming the other methods. However, all methods show pronounced difficulties in correctly categorizing intermediate binders.
由于可能的肽序列数量众多且 MHC 蛋白广泛多态性,因此对大量肽进行 MHC 结合能力的实验筛选仍然非常具有挑战性。因此,开发用于预测肽与 MHC 分子结合能力的计算方法具有重要意义,作为实际筛选中选择肽的第一步。
我们在从免疫表位数据库和分析资源 (IEDB) 中提取的相对较大的 HLA-A 和 HLA-B 等位基因肽结合数据集上,检查了四种不同的 MHC 类 I 预测方法的性能。所选方法涵盖了 MHC 结合预测中可用方法的代表性交叉部分。在 IEDB 开发之前,这种分析是不可能的,因为可用的肽序列数据集很小,分布在许多单独的工作中。我们测试了三个数据集,这些数据集在用于选择结合物和非结合物的 IC50 截止标准上有所不同。当仅对由强结合物(IC50 小于 10 nM)和明确的非结合物(IC50 大于 10,000 nM)组成的数据集进行预测时,获得了最佳性能。此外,只有在具有足够大(大于 200)、平衡的结合物和非结合物集的等位基因上才能实现预测的稳健性。
所有四种方法在综合数据集上均表现出良好到优异的性能,基于人工神经网络的方法表现优于其他方法。然而,所有方法在正确分类中间结合物方面都存在明显的困难。