Institute of Bioinformatics and Systems Biology, National Chiao Tung University, Hsinchu 300, Taiwan.
BMC Bioinformatics. 2010 Feb 24;11:102. doi: 10.1186/1471-2105-11-102.
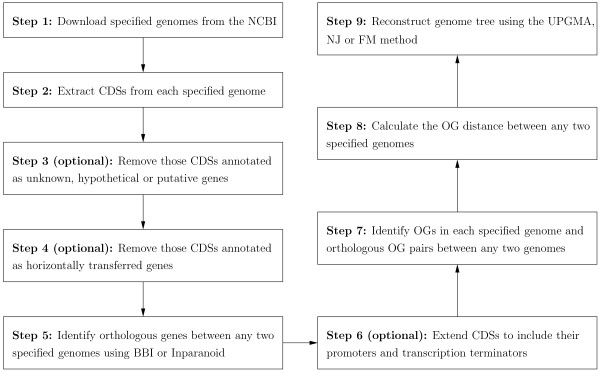
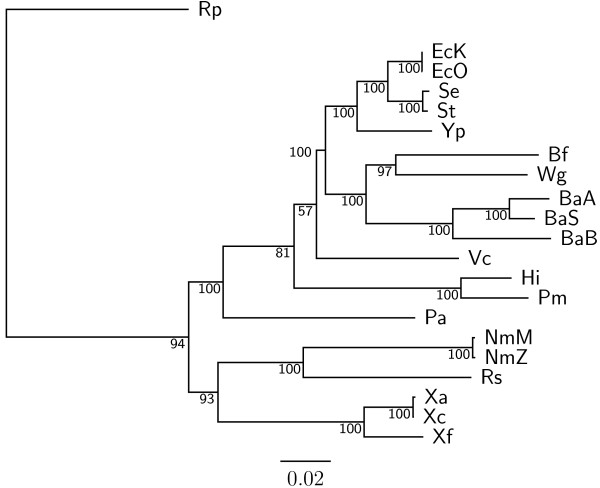
Overlapping genes (OGs) are defined as adjacent genes whose coding sequences overlap partially or entirely. In fact, they are ubiquitous in microbial genomes and more conserved between species than non-overlapping genes. Based on this property, we have previously implemented a web server, named OGtree, that allows the user to reconstruct genome trees of some prokaryotes according to their pairwise OG distances. By analogy to the analyses of gene content and gene order, the OG distance between two genomes we defined was based on a measure of combining OG content (i.e., the normalized number of shared orthologous OG pairs) and OG order (i.e., the normalized OG breakpoint distance) in their whole genomes. A shortcoming of using the concept of breakpoints to define the OG distance is its inability to analyze the OG distance of multi-chromosomal genomes. In addition, the amount of overlapping coding sequences between some distantly related prokaryotic genomes may be limited so that it is hard to find enough OGs to properly evaluate their pairwise OG distances.
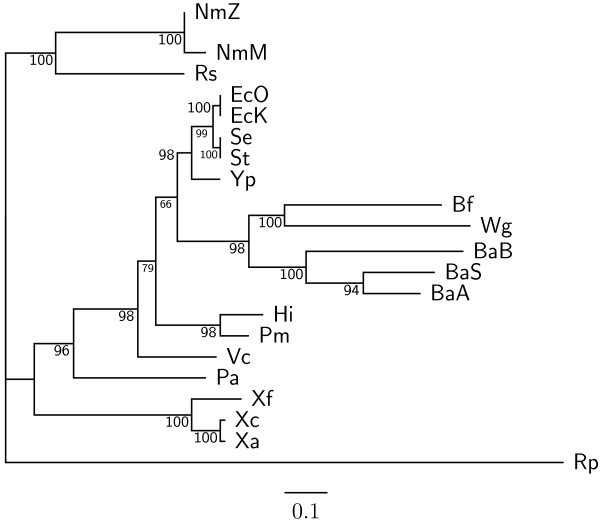
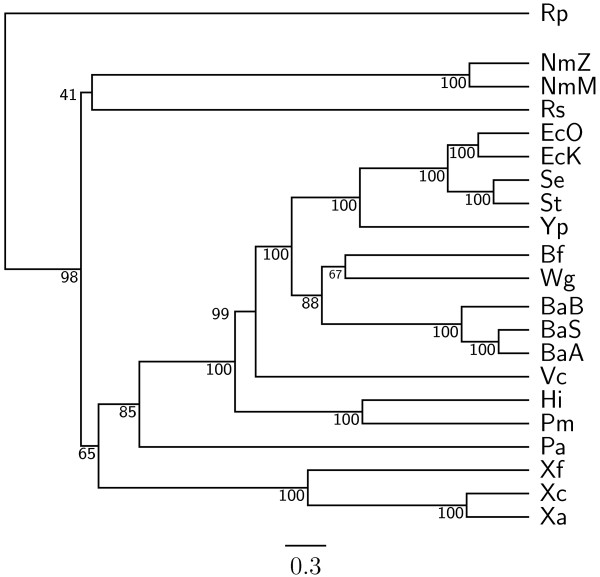
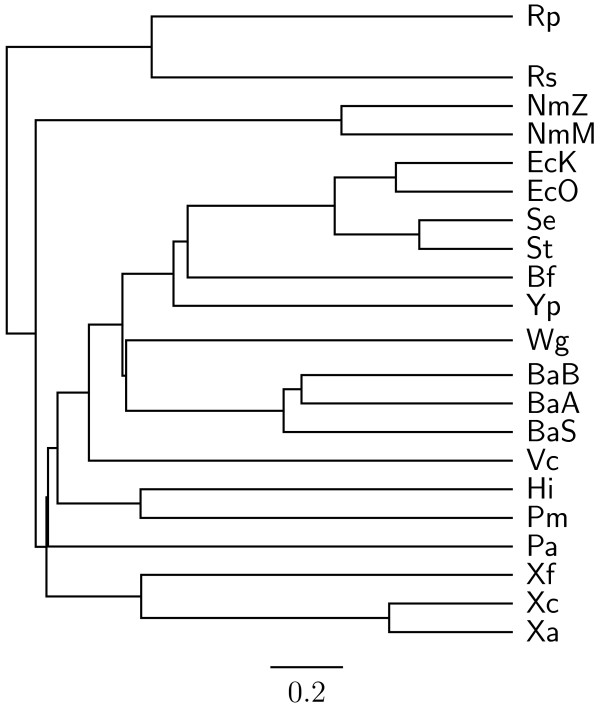
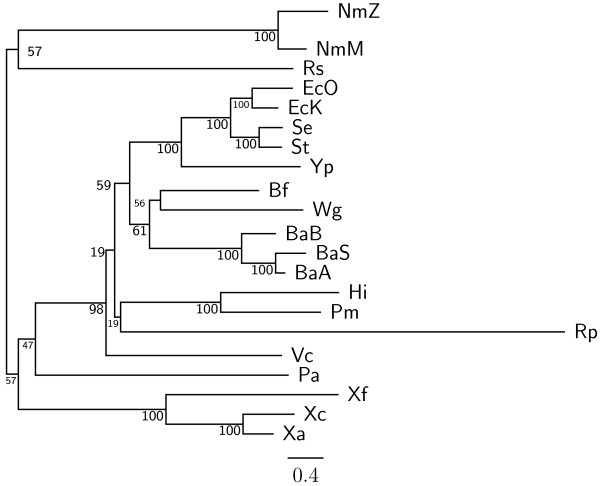
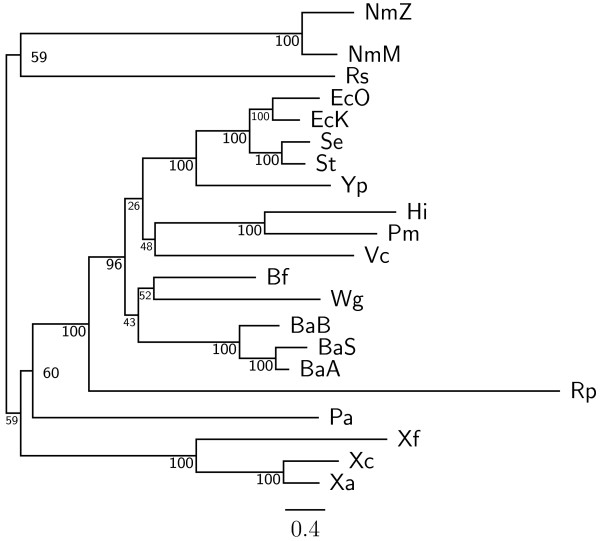
In this study, we therefore define a new OG order distance that is based on more biologically accurate rearrangements (e.g., reversals, transpositions and translocations) rather than breakpoints and that is applicable to both uni-chromosomal and multi-chromosomal genomes. In addition, we expand the term "gene" to include both its coding sequence and regulatory regions so that two adjacent genes whose coding sequences or regulatory regions overlap with each other are considered as a pair of overlapping genes. This is because overlapping of regulatory regions of distinct genes suggests that the regulation of expression for these genes should be more or less interrelated. Based on these modifications, we have reimplemented our OGtree as a new web server, named OGtree2, and have also evaluated its accuracy of genome tree reconstruction on a testing dataset consisting of 21 Proteobacteria genomes. Our experimental results have finally shown that our current OGtree2 indeed outperforms its previous version OGtree, as well as another similar server, called BPhyOG, significantly in the quality of genome tree reconstruction, because the phylogenetic tree obtained by OGtree2 is greatly congruent with the reference tree that coincides with the taxonomy accepted by biologists for these Proteobacteria.
In this study, we have introduced a new web server OGtree2 at http://bioalgorithm.life.nctu.edu.tw/OGtree2.0/ that can serve as a useful tool for reconstructing more precise and robust genome trees of prokaryotes according to their overlapping genes.
重叠基因(OGs)被定义为编码序列部分或完全重叠的相邻基因。事实上,它们在微生物基因组中无处不在,并且在物种间比非重叠基因更保守。基于这一特性,我们之前实现了一个名为 OGtree 的网络服务器,允许用户根据它们的成对 OG 距离来重建某些原核生物的基因组树。通过类比于基因内容和基因顺序的分析,我们定义的两个基因组之间的 OG 距离基于对整个基因组中 OG 内容(即共享直系 OG 对的归一化数量)和 OG 顺序(即归一化 OG 断点距离)的组合的度量。使用断点来定义 OG 距离的一个缺点是它无法分析多染色体基因组的 OG 距离。此外,一些远缘原核生物基因组之间重叠编码序列的数量可能有限,因此很难找到足够的 OG 来正确评估它们的成对 OG 距离。
在这项研究中,我们因此定义了一种新的 OG 顺序距离,该距离基于更准确的生物学重排(例如,反转、转座和易位)而不是断点,并且适用于单染色体和多染色体基因组。此外,我们将“基因”一词扩展到包括其编码序列和调节区域,因此,两个编码序列或调节区域相互重叠的相邻基因被视为一对重叠基因。这是因为不同基因的调节区域重叠表明这些基因的表达调控应该或多或少相互关联。基于这些修改,我们重新实现了我们的 OGtree 作为一个新的网络服务器,命名为 OGtree2,并在一个由 21 个变形菌基因组组成的测试数据集上评估了其重建基因组树的准确性。我们的实验结果最终表明,我们目前的 OGtree2 确实优于其以前的版本 OGtree,以及另一个名为 BPhyOG 的类似服务器,在重建基因组树的质量方面有显著提高,因为 OGtree2 获得的系统发育树与参考树非常一致,该参考树与生物学家为这些变形菌接受的分类学一致。
在这项研究中,我们引入了一个新的网络服务器 OGtree2,位于 http://bioalgorithm.life.nctu.edu.tw/OGtree2.0/,可作为根据重叠基因重建更精确和稳健的原核生物基因组树的有用工具。